Simvastatin
General
Type : Drug || Not A\/B H target || Naphthalen
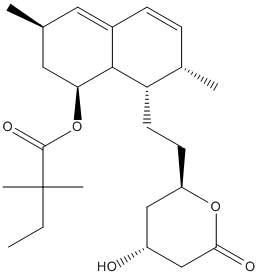
Chemical_Nomenclature : [(1S,3R,7S,8S,8aR)-8-[2-[(2R,4R)-4-hydroxy-6-oxooxan-2-yl]ethyl]-3,7-dimethyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl] 2,2-dimethylbutanoate
Canonical SMILES : CCC(C)(C)C(=O)OC1CC(C=C2C1C(C(C=C2)C)CCC3CC(CC(=O)O3)O)C
InChI : InChI=1S\/C25H38O5\/c1-6-25(4,5)24(28)30-21-12-15(2)11-17-8-7-16(3)20(23(17)21)10-9-19-13-18(26)14-22(27)29-19\/h7-8,11,15-16,18-21,23,26H,6,9-10,12-14H2,1-5H3\/t15-,16-,18+,19+,20-,21-,23-\/m0\/s1
InChIKey : RYMZZMVNJRMUDD-HGQWONQESA-N
Other name(s) : Zocor, Synvinolin, Sinvacor, Denan
Target
Families : Carb_B_Chordata
References (3)
| Title : Role of esterase mediated hydrolysis of simvastatin in human and rat blood and its impact on pharmacokinetic profiles of simvastatin and its active metabolite in rat - Li_2019_J.Pharm.Biomed.Anal_168_13 |
| Author(s) : Li Z , Zhang J , Zhang Y , Zuo Z |
| Ref : J Pharm Biomed Anal , 168 :13 , 2019 |
| Abstract : Li_2019_J.Pharm.Biomed.Anal_168_13 |
| ESTHER : Li_2019_J.Pharm.Biomed.Anal_168_13 |
| PubMedSearch : Li_2019_J.Pharm.Biomed.Anal_168_13 |
| PubMedID: 30776567 |
| Title : Simvastatin requires activation in accessory cells to modulate T-cell responses in asthma and COPD - Knobloch_2016_Eur.J.Pharmacol_788_294 |
| Author(s) : Knobloch J , Yakin Y , Korber S , Grensemann B , Bendella Z , Boyaci N , Gallert WJ , Yanik SD , Jungck D , Koch A |
| Ref : European Journal of Pharmacology , 788 :294 , 2016 |
| Abstract : Knobloch_2016_Eur.J.Pharmacol_788_294 |
| ESTHER : Knobloch_2016_Eur.J.Pharmacol_788_294 |
| PubMedSearch : Knobloch_2016_Eur.J.Pharmacol_788_294 |
| PubMedID: 27343379 |
| Title : Carboxylesterase 1-Mediated Drug-Drug Interactions between Clopidogrel and Simvastatin - Wang_2015_Biol.Pharm.Bull_38_292 |
| Author(s) : Wang X , Zhu HJ , Markowitz JS |
| Ref : Biol Pharm Bull , 38 :292 , 2015 |
| Abstract : Wang_2015_Biol.Pharm.Bull_38_292 |
| ESTHER : Wang_2015_Biol.Pharm.Bull_38_292 |
| PubMedSearch : Wang_2015_Biol.Pharm.Bull_38_292 |
| PubMedID: 25747989 |