Prasugrel
General
Type : Drug || Not A\/B H target || Acetate || Cyclopropane
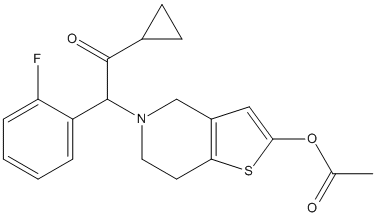
Chemical_Nomenclature : [5-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-6,7-dihydro-4H-thieno[3,2-c]pyridin-2-yl] acetate
Canonical SMILES : CC(=O)OC1=CC2=C(S1)CCN(C2)C(C3=CC=CC=C3F)C(=O)C4CC4
InChI : InChI=1S\/C20H20FNO3S\/c1-12(23)25-18-10-14-11-22(9-8-17(14)26-18)19(20(24)13-6-7-13)15-4-2-3-5-16(15)21\/h2-5,10,13,19H,6-9,11H2,1H3
InChIKey : DTGLZDAWLRGWQN-UHFFFAOYSA-N
Other name(s) : Effient, Efient, CS-747
Target
Families : Carb_B_Chordata, Arylacetamide_deacetylase
References (4)
| Title : Role of human AADAC on hydrolysis of eslicarbazepine acetate and effects of AADAC genetic polymorphisms on hydrolase activity - Hirosawa_2021_Drug.Metab.Dispos__ |
| Author(s) : Hirosawa K , Fukami T , Tashiro K , Sakai Y , Kisui F , Nakano M , Nakajima M |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , : , 2021 |
| Abstract : Hirosawa_2021_Drug.Metab.Dispos__ |
| ESTHER : Hirosawa_2021_Drug.Metab.Dispos__ |
| PubMedSearch : Hirosawa_2021_Drug.Metab.Dispos__ |
| PubMedID: 33446525 |
| Gene_locus related to this paper: human-AADAC |
| Title : Arylacetamide Deacetylase is Responsible for Activation of Prasugrel in Human and Dog - Kurokawa_2016_Drug.Metab.Dispos_44_409 |
| Author(s) : Kurokawa T , Fukami T , Yoshida T , Nakajima M |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 44 :409 , 2016 |
| Abstract : Kurokawa_2016_Drug.Metab.Dispos_44_409 |
| ESTHER : Kurokawa_2016_Drug.Metab.Dispos_44_409 |
| PubMedSearch : Kurokawa_2016_Drug.Metab.Dispos_44_409 |
| PubMedID: 26718653 |
| Title : A comparison of the metabolism of clopidogrel and prasugrel - Laizure_2010_Expert.Opin.Drug.Metab.Toxicol_6_1417 |
| Author(s) : Laizure SC , Parker RB |
| Ref : Expert Opin Drug Metab Toxicol , 6 :1417 , 2010 |
| Abstract : Laizure_2010_Expert.Opin.Drug.Metab.Toxicol_6_1417 |
| ESTHER : Laizure_2010_Expert.Opin.Drug.Metab.Toxicol_6_1417 |
| PubMedSearch : Laizure_2010_Expert.Opin.Drug.Metab.Toxicol_6_1417 |
| PubMedID: 20839914 |
| Title : The biotransformation of prasugrel, a new thienopyridine prodrug, by the human carboxylesterases 1 and 2 - Williams_2008_Drug.Metab.Dispos_36_1227 |
| Author(s) : Williams ET , Jones KO , Ponsler GD , Lowery SM , Perkins EJ , Wrighton SA , Ruterbories KJ , Kazui M , Farid NA |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 36 :1227 , 2008 |
| Abstract : Williams_2008_Drug.Metab.Dispos_36_1227 |
| ESTHER : Williams_2008_Drug.Metab.Dispos_36_1227 |
| PubMedSearch : Williams_2008_Drug.Metab.Dispos_36_1227 |
| PubMedID: 18372401 |