Orlistat
General
Type : Oxooxetan || Lipase inhibitor
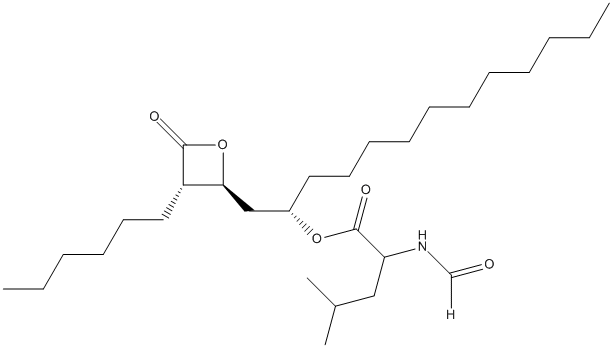
Chemical_Nomenclature : [(2S)-1-[(2S,3S)-3-hexyl-4-oxooxetan-2-yl]tridecan-2-yl] (2S)-2-formamido-4-methylpentanoate
Canonical SMILES : CCCCCCCCCCCC(CC1C(C(=O)O1)CCCCCC)OC(=O)C(CC(C)C)NC=O
InChI : InChI=1S\/C29H53NO5\/c1-5-7-9-11-12-13-14-15-16-18-24(34-29(33)26(30-22-31)20-23(3)4)21-27-25(28(32)35-27)19-17-10-8-6-2\/h22-27H,5-21H2,1-4H3,(H,30,31)\/t24-,25-,26-,27-\/m0\/s1 || InChI=1S\/C29H53NO5\/c1-5-7-9-11-12-13-14-15-16-18-24(34-29(33)26(30-22-31)20-23(3)4)21-27-25(28(32)35-27)19-17-10-8-6-2\/h22-27H,5-21H2,1-4H3,(H,30,31)
InChIKey : AHLBNYSZXLDEJQ-FWEHEUNISA-N || AHLBNYSZXLDEJQ-UHFFFAOYSA-N
Other name(s) : Tetrahydrolipstatin, Orlipastat, Xenical, Alli, (-)-Tetrahydrolipstatin, Orlipastatum, THLP, Lipase Inhibitor, THL, 1-(3-hexyl-4-oxooxetan-2-yl)tridecan-2-yl 2-formamido-4-methylpentanoate, Orlistate, NCGC00095128-01, Orlistat, 5, ACMC-20p1ed
Target
Families : Thioesterase
References (4)
| Title : Total Synthesis of Tetrahydrolipstatin, Its Derivatives, and Evaluation of Their Ability to Potentiate Multiple Antibiotic Classes against Mycobacterium Species - Khan_2021_ACS.Infect.Dis_7_2876 |
| Author(s) : Khan SS , Sudasinghe TD , Landgraf AD , Ronning DR , Sucheck SJ |
| Ref : ACS Infect Dis , 7 :2876 , 2021 |
| Abstract : Khan_2021_ACS.Infect.Dis_7_2876 |
| ESTHER : Khan_2021_ACS.Infect.Dis_7_2876 |
| PubMedSearch : Khan_2021_ACS.Infect.Dis_7_2876 |
| PubMedID: 34478259 |
| Gene_locus related to this paper: myctu-a85c |
| Title : Carboxylesterase-2 is a highly sensitive target of the antiobesity agent orlistat with profound implications in the activation of anticancer prodrugs - Xiao_2013_Biochem.Pharmacol_85_439 |
| Author(s) : Xiao D , Shi D , Yang D , Barthel B , Koch TH , Yan B |
| Ref : Biochemical Pharmacology , 85 :439 , 2013 |
| Abstract : Xiao_2013_Biochem.Pharmacol_85_439 |
| ESTHER : Xiao_2013_Biochem.Pharmacol_85_439 |
| PubMedSearch : Xiao_2013_Biochem.Pharmacol_85_439 |
| PubMedID: 23228697 |
| Gene_locus related to this paper: human-CES2 |
| Title : Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat - Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| Author(s) : Pemble CWt , Johnson LC , Kridel SJ , Lowther WT |
| Ref : Nat Struct Mol Biol , 14 :704 , 2007 |
| Abstract : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| ESTHER : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| PubMedSearch : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| PubMedID: 17618296 |
| Gene_locus related to this paper: human-FASN |
| Title : Orlistat F Hoffmann-La Roche Ltd - Malone_1998_IDrugs_1_232 |
| Author(s) : Malone M |
| Ref : IDrugs , 1 :232 , 1998 |
| Abstract : Malone_1998_IDrugs_1_232 |
| ESTHER : Malone_1998_IDrugs_1_232 |
| PubMedSearch : Malone_1998_IDrugs_1_232 |
| PubMedID: 18465537 |