Methyl-D-lactate
General
Type : Alkyl ester || Propionate
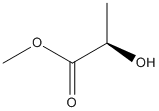
Chemical_Nomenclature : methyl (2R)-2-hydroxypropanoate
Canonical SMILES : CC(C(=O)OC)O
InChI : InChI=1S\/C4H8O3\/c1-3(5)4(6)7-2\/h3,5H,1-2H3\/t3-\/m1\/s1
InChIKey : LPEKGGXMPWTOCB-GSVOUGTGSA-N
Other name(s) : Methyl D-lactate, Methyl (R)-(+)-lactate, (R)-Methyl 2-hydroxypropanoate, Methyl (R)-lactate, (+)-Methyl D-lactate
Target
References (6)
| Title : Characterization of one novel microbial esterase WDEst9 and its use to make l-methyl lactate - Wang_2019_Biocatal.Biotransformation_37_190 |
| Author(s) : Wang Y , Xu S , Li R , Sun A , Zhang Y , Sai K , Hu Y |
| Ref : Biocatalysis and Biotransformation , 37 :190 , 2019 |
| Abstract : Wang_2019_Biocatal.Biotransformation_37_190 |
| ESTHER : Wang_2019_Biocatal.Biotransformation_37_190 |
| PubMedSearch : Wang_2019_Biocatal.Biotransformation_37_190 |
| PubMedID: |
| Gene_locus related to this paper: 9actn-a0a2d1pk72 |
| Title : Functional Characterization of a Marine Bacillus Esterase and its Utilization in the Stereo-Selective Production of D-Methyl Lactate - Huang_2016_Appl.Biochem.Biotechnol_180_1467 |
| Author(s) : Huang J , Zhang Y , Hu Y |
| Ref : Appl Biochem Biotechnol , 180 :1467 , 2016 |
| Abstract : Huang_2016_Appl.Biochem.Biotechnol_180_1467 |
| ESTHER : Huang_2016_Appl.Biochem.Biotechnol_180_1467 |
| PubMedSearch : Huang_2016_Appl.Biochem.Biotechnol_180_1467 |
| PubMedID: 27364331 |
| Gene_locus related to this paper: bacsu-YITV |
| Title : Characterization of a novel marine microbial esterase and its use to make D-methyl lactate - Wang_2016_Chin.J.Catal_37_1396 |
| Author(s) : Wang Y , Zhang Y , Sun A , Hu Y |
| Ref : Chinese Journal of Catalysis , 37 :1396 , 2016 |
| Abstract : Wang_2016_Chin.J.Catal_37_1396 |
| ESTHER : Wang_2016_Chin.J.Catal_37_1396 |
| PubMedSearch : Wang_2016_Chin.J.Catal_37_1396 |
| PubMedID: |
| Gene_locus related to this paper: 9psed-a0a1b1pfx3 |
| Title : Lipase-catalyzed enantioselective synthesis of (R,R)-lactide from alkyl lactate to produce PDLA (poly D-lactic acid) and stereocomplex PLA (poly lactic acid) - Jeon_2013_J.Biotechnol_168_201 |
| Author(s) : Jeon BW , Lee J , Kim HS , Cho DH , Lee H , Chang R , Kim YH |
| Ref : J Biotechnol , 168 :201 , 2013 |
| Abstract : Jeon_2013_J.Biotechnol_168_201 |
| ESTHER : Jeon_2013_J.Biotechnol_168_201 |
| PubMedSearch : Jeon_2013_J.Biotechnol_168_201 |
| PubMedID: 23845270 |
| Gene_locus related to this paper: canar-LipB |
| Title : Lipase-catalyzed oligomerization and hydrolysis of alkyl lactates: direct evidence in the catalysis mechanism that enantioselection is governed by a deacylation step - Ohara_2010_Biomacromolecules_11_2008 |
| Author(s) : Ohara H , Onogi A , Yamamoto M , Kobayashi S |
| Ref : Biomacromolecules , 11 :2008 , 2010 |
| Abstract : Ohara_2010_Biomacromolecules_11_2008 |
| ESTHER : Ohara_2010_Biomacromolecules_11_2008 |
| PubMedSearch : Ohara_2010_Biomacromolecules_11_2008 |
| PubMedID: 20593895 |