Meperidine
General
Type : Piperidine || Opioid analgesic || Drug || Not A\/B H target
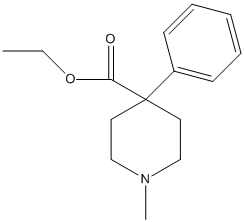
Chemical_Nomenclature : ethyl 1-methyl-4-phenylpiperidine-4-carboxylate
Canonical SMILES : CCOC(=O)C1(CCN(CC1)C)C2=CC=CC=C2
InChI : InChI=1S\/C15H21NO2\/c1-3-18-14(17)15(9-11-16(2)12-10-15)13-7-5-4-6-8-13\/h4-8H,3,9-12H2,1-2H3
InChIKey : XADCESSVHJOZHK-UHFFFAOYSA-N
Other name(s) : Demerol, Pethidine, Meperidine hydrochloride, Meperidine, Isonipecaine, Lidol, Pethanol, Piridosal, Algil, Alodan, Centralgin, Dispadol, Dolantin, Mialgin, Petidin Dolargan, Dolestine, Dolosal, Dolsin, Mefedina
Target
Families : BCHE, Carboxylesterase, Carb_B_Chordata
References (7)
| Title : Selective inhibition of carboxylesterases by isatins, indole-2,3-diones - Hyatt_2007_J.Med.Chem_50_1876 |
| Author(s) : Hyatt JL , Moak T , Hatfield MJ , Tsurkan L , Edwards CC , Wierdl M , Danks MK , Wadkins RM , Potter PM |
| Ref : Journal of Medicinal Chemistry , 50 :1876 , 2007 |
| Abstract : Hyatt_2007_J.Med.Chem_50_1876 |
| ESTHER : Hyatt_2007_J.Med.Chem_50_1876 |
| PubMedSearch : Hyatt_2007_J.Med.Chem_50_1876 |
| PubMedID: 17378546 |
| Title : Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1 - Zhang_1999_J.Pharmacol.Exp.Ther_290_314 |
| Author(s) : Zhang J , Burnell JC , Dumaual N , Bosron WF |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 290 :314 , 1999 |
| Abstract : Zhang_1999_J.Pharmacol.Exp.Ther_290_314 |
| ESTHER : Zhang_1999_J.Pharmacol.Exp.Ther_290_314 |
| PubMedSearch : Zhang_1999_J.Pharmacol.Exp.Ther_290_314 |
| PubMedID: 10381793 |
| Title : Meperidine and alfentanil do not reduce the gain or maximum intensity of shivering - Ikeda_1998_Anesthesiology_88_858 |
| Author(s) : Ikeda T , Sessler DI , Tayefeh F , Negishi C , Turakhia M , Marder D , Bjorksten AR , Larson MD |
| Ref : Anesthesiology , 88 :858 , 1998 |
| Abstract : Ikeda_1998_Anesthesiology_88_858 |
| ESTHER : Ikeda_1998_Anesthesiology_88_858 |
| PubMedSearch : Ikeda_1998_Anesthesiology_88_858 |
| PubMedID: 9579492 |
| Title : Meperidine-induced generalized seizures with normal renal function - Marinella_1997_South.Med.J_90_556 |
| Author(s) : Marinella MA |
| Ref : South Med J , 90 :556 , 1997 |
| Abstract : Marinella_1997_South.Med.J_90_556 |
| ESTHER : Marinella_1997_South.Med.J_90_556 |
| PubMedSearch : Marinella_1997_South.Med.J_90_556 |
| PubMedID: 9160082 |
| Title : Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. - Lockridge_1990_Pharmacol.Ther_47_35 |
| Author(s) : Lockridge O |
| Ref : Pharmacol Ther , 47 :35 , 1990 |
| Abstract : Lockridge_1990_Pharmacol.Ther_47_35 |
| ESTHER : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedSearch : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedID: 2195556 |
| Gene_locus related to this paper: human-BCHE |
| Title : Differential inhibition of plasma cholinesterase variants using the dibutyrate analogue of pancuronium bromide - Whittaker_1981_Hum.Hered_31_242 |
| Author(s) : Whittaker M , Britten JJ |
| Ref : Hum Hered , 31 :242 , 1981 |
| Abstract : Whittaker_1981_Hum.Hered_31_242 |
| ESTHER : Whittaker_1981_Hum.Hered_31_242 |
| PubMedSearch : Whittaker_1981_Hum.Hered_31_242 |
| PubMedID: 7287015 |