Diltiazem
General
Type : Not A\/B H target || Drug || Calcium Channel Antagonist || Azepine || Benzothiazepine
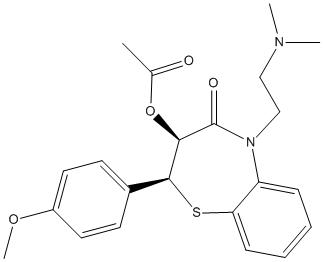
Chemical_Nomenclature : [(2S,3S)-5-[2-(dimethylamino)ethyl]-2-(4-methoxyphenyl)-4-oxo-2,3-dihydro-1,5-benzothiazepin-3-yl] acetate
Canonical SMILES : CC(=O)OC1C(SC2=CC=CC=C2N(C1=O)CCN(C)C)C3=CC=C(C=C3)OC
InChI : InChI=1S\/C22H26N2O4S\/c1-15(25)28-20-21(16-9-11-17(27-4)12-10-16)29-19-8-6-5-7-18(19)24(22(20)26)14-13-23(2)3\/h5-12,20-21H,13-14H2,1-4H3\/t20-,21+\/m1\/s1
InChIKey : HSUGRBWQSSZJOP-RTWAWAEBSA-N
Other name(s) : d-cis-Diltiazem, Cardizem, Dilt-cd
Target
Families : Carb_B_Chordata
References (9)
| Title : Structure of a gut microbial diltiazem-metabolizing enzyme suggests possible substrate binding mode - Zhou_2020_Biochem.Biophys.Res.Commun__ |
| Author(s) : Zhou S , Ko TP , Huang JW , Liu W , Zheng Y , Wu S , Wang Q , Xie Z , Liu Z , Chen CC , Guo RT |
| Ref : Biochemical & Biophysical Research Communications , : , 2020 |
| Abstract : Zhou_2020_Biochem.Biophys.Res.Commun__ |
| ESTHER : Zhou_2020_Biochem.Biophys.Res.Commun__ |
| PubMedSearch : Zhou_2020_Biochem.Biophys.Res.Commun__ |
| PubMedID: 32423809 |
| Title : The P-glycoprotein inhibitor diltiazem-like 8-(4-chlorophenyl)-5-methyl-8-[(2Z)-pent-2-en-1-yloxy]-8H-[1,2,4]oxadiazolo[3,4-c ][1,4]thiazin-3-one inhibits esterase activity and H3 histone acetylation - Cuviello_2018_Eur.J.Med.Chem_164_1 |
| Author(s) : Cuviello F , Bisaccia F , Spinelli D , Armentano MF , Bufo SA , Cosimelli B , Ostuni A |
| Ref : Eur Journal of Medicinal Chemistry , 164 :1 , 2018 |
| Abstract : Cuviello_2018_Eur.J.Med.Chem_164_1 |
| ESTHER : Cuviello_2018_Eur.J.Med.Chem_164_1 |
| PubMedSearch : Cuviello_2018_Eur.J.Med.Chem_164_1 |
| PubMedID: 30583246 |
| Title : Characterization of Species Differences in Tissue Diltiazem Deacetylation Identifies Ces2a as a Rat-Specific Diltiazem Deacetylase - Kurokawa_2015_Drug.Metab.Dispos_43_1218 |
| Author(s) : Kurokawa T , Fukami T , Nakajima M |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 43 :1218 , 2015 |
| Abstract : Kurokawa_2015_Drug.Metab.Dispos_43_1218 |
| ESTHER : Kurokawa_2015_Drug.Metab.Dispos_43_1218 |
| PubMedSearch : Kurokawa_2015_Drug.Metab.Dispos_43_1218 |
| PubMedID: 25979260 |
| Title : Protective effect of a calcium channel blocker diltiazem on aluminum chloride-induced dementia in mice - Rani_2015_Naunyn.Schmiedebergs.Arch.Pharmacol_388_1151 |
| Author(s) : Rani A , Neha , Sodhi RK , Kaur A |
| Ref : Naunyn Schmiedebergs Arch Pharmacol , 388 :1151 , 2015 |
| Abstract : Rani_2015_Naunyn.Schmiedebergs.Arch.Pharmacol_388_1151 |
| ESTHER : Rani_2015_Naunyn.Schmiedebergs.Arch.Pharmacol_388_1151 |
| PubMedSearch : Rani_2015_Naunyn.Schmiedebergs.Arch.Pharmacol_388_1151 |
| PubMedID: 26142889 |
| Title : Significantly improved expression and biochemical properties of recombinant Serratia marcescens lipase as robust biocatalyst for kinetic resolution of chiral ester - Wang_2010_Appl.Biochem.Biotechnol_162_2387 |
| Author(s) : Wang Y , Zhao J , Xu JH , Fan LQ , Li SX , Zhao LL , Mao XB |
| Ref : Appl Biochem Biotechnol , 162 :2387 , 2010 |
| Abstract : Wang_2010_Appl.Biochem.Biotechnol_162_2387 |
| ESTHER : Wang_2010_Appl.Biochem.Biotechnol_162_2387 |
| PubMedSearch : Wang_2010_Appl.Biochem.Biotechnol_162_2387 |
| PubMedID: 20574813 |
| Title : Effects of diltiazem on human nicotinic acetylcholine and GABA(A) receptors - Houlihan_2000_Neuropharmacol_39_2533 |
| Author(s) : Houlihan LM , Slater EY , Beadle DJ , Lukas RJ , Bermudez I |
| Ref : Neuropharmacology , 39 :2533 , 2000 |
| Abstract : Houlihan_2000_Neuropharmacol_39_2533 |
| ESTHER : Houlihan_2000_Neuropharmacol_39_2533 |
| PubMedSearch : Houlihan_2000_Neuropharmacol_39_2533 |
| PubMedID: 11044725 |
| Title : Lipases: Interfacial Enzymes with Attractive Applications - Schmid_1998_Angew.Chem.Int.Ed.Engl_37_1608 |
| Author(s) : Schmid RD , Verger R |
| Ref : Angew Chem Int Ed Engl , 37 :1608 , 1998 |
| Abstract : Schmid_1998_Angew.Chem.Int.Ed.Engl_37_1608 |
| ESTHER : Schmid_1998_Angew.Chem.Int.Ed.Engl_37_1608 |
| PubMedSearch : Schmid_1998_Angew.Chem.Int.Ed.Engl_37_1608 |
| PubMedID: 29711530 |
| Title : Effect of calcium antagonists (omega-conotoxin GVIA, verapamil, gallopamil, diltiazem) on bronchial smooth muscle contractions induced by soman - Walday_1992_Naunyn.Schmiedebergs.Arch.Pharmacol_346_352 |
| Author(s) : Walday P , Fyllingen E , Aas P |
| Ref : Naunyn Schmiedebergs Arch Pharmacol , 346 :352 , 1992 |
| Abstract : Walday_1992_Naunyn.Schmiedebergs.Arch.Pharmacol_346_352 |
| ESTHER : Walday_1992_Naunyn.Schmiedebergs.Arch.Pharmacol_346_352 |
| PubMedSearch : Walday_1992_Naunyn.Schmiedebergs.Arch.Pharmacol_346_352 |
| PubMedID: 1407018 |
| Title : Calcium channel blocker reverses anticholinesterase-induced myopathy - Meshul_1989_Brain.Res_497_142 |
| Author(s) : Meshul CK |
| Ref : Brain Research , 497 :142 , 1989 |
| Abstract : Meshul_1989_Brain.Res_497_142 |
| ESTHER : Meshul_1989_Brain.Res_497_142 |
| PubMedSearch : Meshul_1989_Brain.Res_497_142 |
| PubMedID: 2790449 |