Capecitabine
General
Type : Drug || Carbamate || Pyrimidine || Pro-Drug
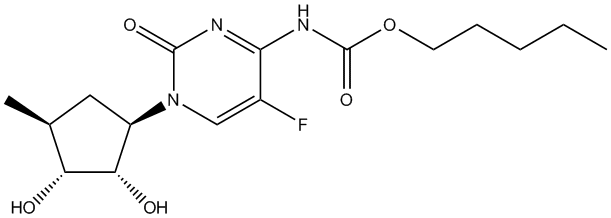
Chemical_Nomenclature : pentyl N-[1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-methyloxolan-2-yl]-5-fluoro-2-oxopyrimidin-4-yl]carbamate
Canonical SMILES : CCCCCOC(=O)NC1=NC(=O)N(C=C1F)C2C(C(C(O2)C)O)O
InChI : InChI=1S\/C15H22FN3O6\/c1-3-4-5-6-24-15(23)18-12-9(16)7-19(14(22)17-12)13-11(21)10(20)8(2)25-13\/h7-8,10-11,13,20-21H,3-6H2,1-2H3,(H,17,18,22,23)\/t8-,10-,11-,13-\/m1\/s1
InChIKey : GAGWJHPBXLXJQN-UORFTKCHSA-N
Other name(s) : Xeloda, capecitabina, capecitabinum, Capecitabin, Capecitibine, Capiibine, Caxeta, Xabine
Target
Families : Carboxylesterase, Carb_B_Chordata
References (16)
| Title : Capecitabine-induced hand-foot syndrome: A pharmacogenetic study beyond DPYD - de With_2023_Biomed.Pharmacother_159_114232 |
| Author(s) : de With M , van Doorn L , Maasland DC , Mulder TAM , Oomen-de Hoop E , Mostert B , Homs MYV , El Bouazzaoui S , Mathijssen RHJ , van Schaik RHN , Bins S |
| Ref : Biomed Pharmacother , 159 :114232 , 2023 |
| Abstract : de With_2023_Biomed.Pharmacother_159_114232 |
| ESTHER : de With_2023_Biomed.Pharmacother_159_114232 |
| PubMedSearch : de With_2023_Biomed.Pharmacother_159_114232 |
| PubMedID: 36630849 |
| Title : Assessment of drug-drug interaction and optimization in capecitabine and irinotecan combination regimen using a physiologically based pharmacokinetic model - Sakai_2021_J.Pharm.Sci__ |
| Author(s) : Sakai S , Kobuchi S , Ito Y , Sakaeda T |
| Ref : J Pharm Sci , : , 2021 |
| Abstract : Sakai_2021_J.Pharm.Sci__ |
| ESTHER : Sakai_2021_J.Pharm.Sci__ |
| PubMedSearch : Sakai_2021_J.Pharm.Sci__ |
| PubMedID: 34965386 |
| Title : Novel Genetic Variants in Carboxylesterase 1 Predict Severe Early-Onset Capecitabine-Related Toxicity - Hamzic_2017_Clin.Pharmacol.Ther_102_796 |
| Author(s) : Hamzic S , Kummer D , Milesi S , Mueller D , Joerger M , Aebi S , Amstutz U , Largiader CR |
| Ref : Clinical Pharmacology & Therapeutics , 102 :796 , 2017 |
| Abstract : Hamzic_2017_Clin.Pharmacol.Ther_102_796 |
| ESTHER : Hamzic_2017_Clin.Pharmacol.Ther_102_796 |
| PubMedSearch : Hamzic_2017_Clin.Pharmacol.Ther_102_796 |
| PubMedID: 28139840 |
| Gene_locus related to this paper: human-CES1 |
| Title : A Simple Method for Comparing Enzymatic Capecitabine Activation in Various Mono- and Combination Chemotherapies - Buchner_2015_Pharmacology_95_29 |
| Author(s) : Buchner P , Sahmanovic A , Schreiber V , Baroian N , Dittrich C , Czejka M |
| Ref : Pharmacology , 95 :29 , 2015 |
| Abstract : Buchner_2015_Pharmacology_95_29 |
| ESTHER : Buchner_2015_Pharmacology_95_29 |
| PubMedSearch : Buchner_2015_Pharmacology_95_29 |
| PubMedID: 25591914 |
| Title : Standard versus continuous administration of capecitabine in metastatic breast cancer (GEICAM\/2009-05): a randomized, noninferiority phase II trial with a pharmacogenetic analysis - Martin_2015_Oncologist_20_111 |
| Author(s) : Martin M , Martinez N , Ramos M , Calvo L , Lluch A , Zamora P , Munoz M , Carrasco E , Caballero R , Garcia-Saenz JA , Guerra E , Caronia D , Casado A , Ruiz-Borrego M , Hernando B , Chacon JI , De la Torre-Montero JC , Jimeno MA , Heras L , Alonso R , De la Haba J , Pita G , Constenla M , Gonzalez-Neira A |
| Ref : Oncologist , 20 :111 , 2015 |
| Abstract : Martin_2015_Oncologist_20_111 |
| ESTHER : Martin_2015_Oncologist_20_111 |
| PubMedSearch : Martin_2015_Oncologist_20_111 |
| PubMedID: 25601966 |
| Title : ZRX1, the first EGFR inhibitor-capecitabine based combi-molecule, requires carboxylesterase-mediated hydrolysis for optimal activity - Ait-Tihyaty_2013_Invest.New.Drugs_31_1409 |
| Author(s) : Ait-Tihyaty M , Rachid Z , Larroque-Lombard AL , Jean-Claude BJ |
| Ref : Invest New Drugs , 31 :1409 , 2013 |
| Abstract : Ait-Tihyaty_2013_Invest.New.Drugs_31_1409 |
| ESTHER : Ait-Tihyaty_2013_Invest.New.Drugs_31_1409 |
| PubMedSearch : Ait-Tihyaty_2013_Invest.New.Drugs_31_1409 |
| PubMedID: 23959266 |
| Title : Higher capecitabine AUC in elderly patients with advanced colorectal cancer (SWOGS0030) - Louie_2013_Br.J.Cancer_109_1744 |
| Author(s) : Louie SG , Ely B , Lenz HJ , Albain KS , Gotay C , Coleman D , Raghavan D , Shields AF , Gold PJ , Blanke CD |
| Ref : Br J Cancer , 109 :1744 , 2013 |
| Abstract : Louie_2013_Br.J.Cancer_109_1744 |
| ESTHER : Louie_2013_Br.J.Cancer_109_1744 |
| PubMedSearch : Louie_2013_Br.J.Cancer_109_1744 |
| PubMedID: 24022189 |
| Title : Phase II and gene expression analysis trial of neoadjuvant capecitabine plus irinotecan followed by capecitabine-based chemoradiotherapy for locally advanced rectal cancer: Hoosier Oncology Group GI03-53 - Chiorean_2012_Cancer.Chemother.Pharmacol_70_25 |
| Author(s) : Chiorean EG , Sanghani S , Schiel MA , Yu M , Burns M , Tong Y , Hinkle DT , Coleman N , Robb B , LeBlanc J , Clark R , Bufill J , Curie C , Loehrer PJ , Cardenes H |
| Ref : Cancer Chemother Pharmacol , 70 :25 , 2012 |
| Abstract : Chiorean_2012_Cancer.Chemother.Pharmacol_70_25 |
| ESTHER : Chiorean_2012_Cancer.Chemother.Pharmacol_70_25 |
| PubMedSearch : Chiorean_2012_Cancer.Chemother.Pharmacol_70_25 |
| PubMedID: 22610353 |
| Title : A polymorphism in the cytidine deaminase promoter predicts severe capecitabine-induced hand-foot syndrome - Caronia_2011_Clin.Cancer.Res_17_2006 |
| Author(s) : Caronia D , Martin M , Sastre J , de la Torre J , Garcia-Saenz JA , Alonso MR , Moreno LT , Pita G , Diaz-Rubio E , Benitez J , Gonzalez-Neira A |
| Ref : Clin Cancer Research , 17 :2006 , 2011 |
| Abstract : Caronia_2011_Clin.Cancer.Res_17_2006 |
| ESTHER : Caronia_2011_Clin.Cancer.Res_17_2006 |
| PubMedSearch : Caronia_2011_Clin.Cancer.Res_17_2006 |
| PubMedID: 21325291 |
| Title : Capecitabine, oxaliplatin and radiotherapy: a phase IB neoadjuvant study for esophageal cancer with gene expression analysis - Javle_2009_Cancer.Invest_27_193 |
| Author(s) : Javle MM , Yang G , Nwogu CE , Wilding GE , O'Malley L , Vinjamaram S , Schiff MD , Nava HR , LeVea C , Clark KR , Prey JD , Smith PF , Pendyala L |
| Ref : Cancer Invest , 27 :193 , 2009 |
| Abstract : Javle_2009_Cancer.Invest_27_193 |
| ESTHER : Javle_2009_Cancer.Invest_27_193 |
| PubMedSearch : Javle_2009_Cancer.Invest_27_193 |
| PubMedID: 19235592 |
| Title : A carboxylesterase 2 gene polymorphism as predictor of capecitabine on response and time to progression - Ribelles_2008_Curr.Drug.Metab_9_336 |
| Author(s) : Ribelles N , Lopez-Siles J , Sanchez A , Gonzalez E , Sanchez MJ , Carabantes F , Sanchez-Rovira P , Marquez A , Duenas R , Sevilla I , Alba E |
| Ref : Curr Drug Metab , 9 :336 , 2008 |
| Abstract : Ribelles_2008_Curr.Drug.Metab_9_336 |
| ESTHER : Ribelles_2008_Curr.Drug.Metab_9_336 |
| PubMedSearch : Ribelles_2008_Curr.Drug.Metab_9_336 |
| PubMedID: 18473752 |
| Title : Capecitabine-induced severe hypertriglyceridemia: report of two cases - Kurt_2006_Ann.Pharmacother_40_328 |
| Author(s) : Kurt M , Babaoglu MO , Yasar U , Shorbagi A , Guler N |
| Ref : Annals of Pharmacotherapy , 40 :328 , 2006 |
| Abstract : Kurt_2006_Ann.Pharmacother_40_328 |
| ESTHER : Kurt_2006_Ann.Pharmacother_40_328 |
| PubMedSearch : Kurt_2006_Ann.Pharmacother_40_328 |
| PubMedID: 16391007 |
| Title : Mammalian carboxylesterases: from drug targets to protein therapeutics - Redinbo_2005_Drug.Discov.Today_10_313 |
| Author(s) : Redinbo MR , Potter PM |
| Ref : Drug Discov Today , 10 :313 , 2005 |
| Abstract : Redinbo_2005_Drug.Discov.Today_10_313 |
| ESTHER : Redinbo_2005_Drug.Discov.Today_10_313 |
| PubMedSearch : Redinbo_2005_Drug.Discov.Today_10_313 |
| PubMedID: 15749280 |
| Title : Hydrolysis of capecitabine to 5'-deoxy-5-fluorocytidine by human carboxylesterases and inhibition by loperamide - Quinney_2005_J.Pharmacol.Exp.Ther_313_1011 |
| Author(s) : Quinney SK , Sanghani SP , Davis WI , Hurley TD , Sun Z , Murry DJ , Bosron WF |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 313 :1011 , 2005 |
| Abstract : Quinney_2005_J.Pharmacol.Exp.Ther_313_1011 |
| ESTHER : Quinney_2005_J.Pharmacol.Exp.Ther_313_1011 |
| PubMedSearch : Quinney_2005_J.Pharmacol.Exp.Ther_313_1011 |
| PubMedID: 15687373 |
| Gene_locus related to this paper: human-CES1 , human-CES2 , human-CES3 |
| Title : Capecitabine in the treatment of metastatic renal cell carcinoma failing immunotherapy - Wenzel_2002_Am.J.Kidney.Dis_39_48 |
| Author(s) : Wenzel C , Locker GJ , Schmidinger M , Mader R , Kramer G , Marberger M , Rauchenwald M , Zielinski CC , Steger GG |
| Ref : Am J Kidney Dis , 39 :48 , 2002 |
| Abstract : Wenzel_2002_Am.J.Kidney.Dis_39_48 |
| ESTHER : Wenzel_2002_Am.J.Kidney.Dis_39_48 |
| PubMedSearch : Wenzel_2002_Am.J.Kidney.Dis_39_48 |
| PubMedID: 11774101 |
| Title : Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue - Miwa_1998_Eur.J.Cancer_34_1274 |
| Author(s) : Miwa M , Ura M , Nishida M , Sawada N , Ishikawa T , Mori K , Shimma N , Umeda I , Ishitsuka H |
| Ref : Eur J Cancer , 34 :1274 , 1998 |
| Abstract : Miwa_1998_Eur.J.Cancer_34_1274 |
| ESTHER : Miwa_1998_Eur.J.Cancer_34_1274 |
| PubMedSearch : Miwa_1998_Eur.J.Cancer_34_1274 |
| PubMedID: 9849491 |