N-acetylanthranilate
General
Type : Natural || Benzoate
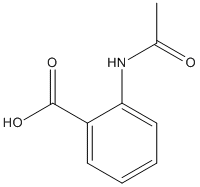
Chemical_Nomenclature : 2-acetamidobenzoic acid
Canonical SMILES : CC(=O)NC1=CC=CC=C1C(=O)O
InChI : InChI=1S\/C9H9NO3\/c1-6(11)10-8-5-3-2-4-7(8)9(12)13\/h2-5H,1H3,(H,10,11)(H,12,13)
InChIKey : QSACCXVHEVWNMX-UHFFFAOYSA-N
Other name(s) : 2-Acetamidobenzoic acid, N-ACETYLANTHRANILIC ACID, 2-(Acetylamino)benzoic acid, 2-Carboxyacetanilide, Acetylanthranilic acid, ZZ8, 2-acetamidobenzoate, Acetamidobenzoate, P-acetaminobenzoate, 2-(acetylamino)benzoate, AC1NUSR8
Target
References (4)
| Title : Evolutionary adaptation from hydrolytic to oxygenolytic catalysis at the alpha\/beta fold - Bui_2023_Chem.Sci_14_10547 |
| Author(s) : Bui S , Gil-Guerrero S , van der Linden P , Carpentier P , Ceccarelli M , Jambrina PG , Steiner RA |
| Ref : Chem Sci , 14 :10547 , 2023 |
| Abstract : Bui_2023_Chem.Sci_14_10547 |
| ESTHER : Bui_2023_Chem.Sci_14_10547 |
| PubMedSearch : Bui_2023_Chem.Sci_14_10547 |
| PubMedID: 37799987 |
| Gene_locus related to this paper: artsp-hod |
| Title : Structural basis for cofactor-independent dioxygenation of N-heteroaromatic compounds at the alpha\/beta-hydrolase fold - Steiner_2010_Proc.Natl.Acad.Sci.U.S.A_107_657 |
| Author(s) : Steiner RA , Janssen HJ , Roversi P , Oakley AJ , Fetzner S |
| Ref : Proc Natl Acad Sci U S A , 107 :657 , 2010 |
| Abstract : Steiner_2010_Proc.Natl.Acad.Sci.U.S.A_107_657 |
| ESTHER : Steiner_2010_Proc.Natl.Acad.Sci.U.S.A_107_657 |
| PubMedSearch : Steiner_2010_Proc.Natl.Acad.Sci.U.S.A_107_657 |
| PubMedID: 20080731 |
| Gene_locus related to this paper: artsp-hod , psepu-QDO |
| Title : N-acetylanthranilate amidase from Arthrobacter nitroguajacolicus Ru61a, an alpha\/beta-hydrolase-fold protein active towards aryl-acylamides and -esters, and properties of its cysteine-deficient variant - Kolkenbrock_2006_J.Bacteriol_188_8430 |
| Author(s) : Kolkenbrock S , Parschat K , Beermann B , Hinz HJ , Fetzner S |
| Ref : Journal of Bacteriology , 188 :8430 , 2006 |
| Abstract : Kolkenbrock_2006_J.Bacteriol_188_8430 |
| ESTHER : Kolkenbrock_2006_J.Bacteriol_188_8430 |
| PubMedSearch : Kolkenbrock_2006_J.Bacteriol_188_8430 |
| PubMedID: 17041061 |
| Gene_locus related to this paper: 9micc-q7wsq8 |
| Title : 2,4-dioxygenases catalyzing N-heterocyclic-ring cleavage and formation of carbon monoxide. Purification and some properties of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase from Arthrobacter sp. Ru61a and comparison with 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase from Pseudomonas putida 33\/1 - Bauer_1996_Eur.J.Biochem_240_576 |
| Author(s) : Bauer I , Max N , Fetzner S , Lingens F |
| Ref : European Journal of Biochemistry , 240 :576 , 1996 |
| Abstract : Bauer_1996_Eur.J.Biochem_240_576 |
| ESTHER : Bauer_1996_Eur.J.Biochem_240_576 |
| PubMedSearch : Bauer_1996_Eur.J.Biochem_240_576 |
| PubMedID: 8856057 |
| Gene_locus related to this paper: artsp-hod , psepu-QDO |