SAD-128
General
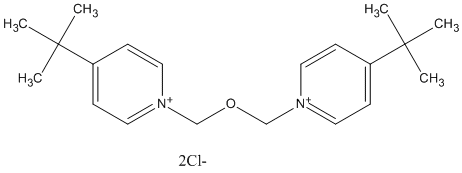
Type : Bispyridinium, non-oxime, tert-Butyl
Chemical_Nomenclature : 1-tert-butyl-4-[(4-tert-butylpyridin-1-ium-1-yl)methoxymethyl]pyridin-1-ium\;dichloride
Canonical SMILES : CC(C)(C)C1=CC=[N+](C=C1)COCC2=CC=[N+](C=C2)C(C)(C)C.[Cl-].[Cl-]
InChI : InChI=1S\/C20H30N2O.2ClH\/c1-19(2,3)18-9-11-21(12-10-18)16-23-15-17-7-13-22(14-8-17)20(4,5)6\;\;\/h7-14H,15-16H2,1-6H3\;2*1H\/q+2\;\;\/p-2
InChIKey : WZPZUOXQRQHKKA-UHFFFAOYSA-L
Other name(s) : 4-tert-butyl-1-{[(1-tert-butylpyridinium-4-yl)methoxy]methyl}pyridinium dichloride, Pyridinium, 1,1'-oxydimethylenebis(4-tert-butyl-, dichloride, 1,1'-(Oxybis(methylene))bis(4-(1,1-dimethylethyl)pyridinium), dichloride, Bis-(4-t-butylpyridine)-1-methylether dichloride, 1,1'-Oxydimethylene bis(4-tert-butylpyridinium chloride)
MW : 385.371
Formula : C20H30Cl2N2O
CAS_number :
CID PubChem :
InChIKey UniChem :
Iuphar :
Wikipedia :
Target
Structure : No structure
Families : No family
References
No reference