RS194B
General
Type : Uncharged Oxime, Oxime, Azepane
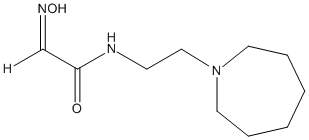
Chemical_Nomenclature : N-[2-(azepan-1-yl)ethyl]-2-hydroxyiminoacetamide
Canonical SMILES : C1CCCN(CC1)CCNC(=O)C=NO
InChI : InChI=1S\/C10H19N3O2\/c14-10(9-12-15)11-5-8-13-6-3-1-2-4-7-13\/h9,15H,1-8H2,(H,11,14)
InChIKey : VIABFASHQHHYNB-UHFFFAOYSA-N
Other name(s) : RS-194B, AC1OAKUK, Oprea1_728133, SCHEMBL15963820, MCULE-3450389482, 2-(Hydroxyimino)-iV-(2-(azepan-l-yl)ethyl)acetamide
MW : 214.5
Formula : C10H19N3O2
CAS_number :
CID PubChem :
InChIKey UniChem :
Iuphar :
Wikipedia :
References
No reference