t-AUCB
General
Type : Adamantyl,Urea derivative
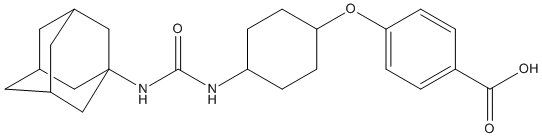
Chemical_Nomenclature : 4-[4-(1-adamantylcarbamoylamino)cyclohexyl]oxybenzoic acid
Canonical SMILES : C1CC(CCC1NC(=O)NC23CC4CC(C2)CC(C4)C3)OC5=CC=C(C=C5)C(=O)O
InChI : InChI=1S\/C24H32N2O4\/c27-22(28)18-1-5-20(6-2-18)30-21-7-3-19(4-8-21)25-23(29)26-24-12-15-9-16(13-24)11-17(10-15)14-24\/h1-2,5-6,15-17,19,21H,3-4,7-14H2,(H,27,28)(H2,25,26,29)
InChIKey : KNBWKJBQDAQARU-UHFFFAOYSA-N
Other name(s) : CHEMBL242459,CHEMBL244405,Urea-based compound, 21,AUB,3wke,Trans-ACUB,4-[(Trans-4-{[(3s,5s,7s)-Tricyclo[3.3.1.1~3,7~]dec-1-Ylcarbamoyl]amino}cyclohexyl)oxy]benzoic Acid
MW : 412.53
Formula : C24H32N2O4
CAS_number :
PubChem : 16038368
UniChem : KNBWKJBQDAQARU-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : t-AUCB ligand of proteins in family: Epoxide_hydrolase
Stucture :
Protein : human-EPHX2 || hypjq-g0r7e2
References (10)
| Title : Soluble Epoxide Hydrolase Inhibitor t-AUCB Promotes Murine Brown Adipogenesis: Role of PPAR Gamma and PPAR Alpha (P21-069-19) - Hildreth_2019_Curr.Dev.Nutr_3_ |
| Author(s) : Hildreth K , Overby H , Kodani S , Morisseau C , Hammock B , Bettaieb A , Zhao L |
| Ref : Curr Dev Nutr , 3 : , 2019 |
| Abstract : Hildreth_2019_Curr.Dev.Nutr_3_ |
| ESTHER : Hildreth_2019_Curr.Dev.Nutr_3_ |
| PubMedSearch : Hildreth_2019_Curr.Dev.Nutr_3_ |
| PubMedID: 31223951 |
| Title : The molecular structure of an epoxide hydrolase from Trichoderma reesei in complex with urea or amide-based inhibitors - de Oliveira_2019_Int.J.Biol.Macromol_129_653 |
| Author(s) : de Oliveira GS , Adriani PP , Ribeiro JA , Morisseau C , Hammock BD , Dias MVB , Chambergo FS |
| Ref : Int J Biol Macromol , 129 :653 , 2019 |
| Abstract : de Oliveira_2019_Int.J.Biol.Macromol_129_653 |
| ESTHER : de Oliveira_2019_Int.J.Biol.Macromol_129_653 |
| PubMedSearch : de Oliveira_2019_Int.J.Biol.Macromol_129_653 |
| PubMedID: 30771398 |
| Gene_locus related to this paper: hypjq-g0r7e2 |
| Title : Substrate and inhibitor selectivity, and biological activity of an epoxide hydrolase from Trichoderma reesei - de Oliveira_2019_Mol.Biol.Rep_46_371 |
| Author(s) : de Oliveira GS , Adriani PP , Wu H , Morisseau C , Hammock BD , Chambergo FS |
| Ref : Mol Biol Rep , 46 :371 , 2019 |
| Abstract : de Oliveira_2019_Mol.Biol.Rep_46_371 |
| ESTHER : de Oliveira_2019_Mol.Biol.Rep_46_371 |
| PubMedSearch : de Oliveira_2019_Mol.Biol.Rep_46_371 |
| PubMedID: 30426381 |
| Gene_locus related to this paper: hypjq-g0r7e2 |
| Title : Soluble epoxide hydrolase inhibitors, t-AUCB, downregulated miR-133 in a mouse model of myocardial infarction - Gui_2018_Lipids.Health.Dis_17_129 |
| Author(s) : Gui Y , Li D , Chen J , Wang Y , Hu J , Liao C , Deng L , Xiang Q , Yang T , Du X , Zhang S , Xu D |
| Ref : Lipids Health Dis , 17 :129 , 2018 |
| Abstract : Gui_2018_Lipids.Health.Dis_17_129 |
| ESTHER : Gui_2018_Lipids.Health.Dis_17_129 |
| PubMedSearch : Gui_2018_Lipids.Health.Dis_17_129 |
| PubMedID: 29843720 |
| Title : Successful generation of structural information for fragment-based drug discovery - Oster_2015_Drug.Discov.Today_20_1104 |
| Author(s) : Oster L , Tapani S , Xue Y , Kack H |
| Ref : Drug Discov Today , 20 :1104 , 2015 |
| Abstract : Oster_2015_Drug.Discov.Today_20_1104 |
| ESTHER : Oster_2015_Drug.Discov.Today_20_1104 |
| PubMedSearch : Oster_2015_Drug.Discov.Today_20_1104 |
| PubMedID: 25931264 |
| Gene_locus related to this paper: human-EPHX2 |
| Title : Structural insights into binding of inhibitors to soluble epoxide hydrolase gained by fragment screening and X-ray crystallography - Amano_2014_Bioorg.Med.Chem_22_2427 |
| Author(s) : Amano Y , Yamaguchi T , Tanabe E |
| Ref : Bioorganic & Medicinal Chemistry , 22 :2427 , 2014 |
| Abstract : Amano_2014_Bioorg.Med.Chem_22_2427 |
| ESTHER : Amano_2014_Bioorg.Med.Chem_22_2427 |
| PubMedSearch : Amano_2014_Bioorg.Med.Chem_22_2427 |
| PubMedID: 24656800 |
| Gene_locus related to this paper: human-EPHX2 |
| Title : A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARgamma to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction - Xu_2013_Int.J.Cardiol_167_1298 |
| Author(s) : Xu DY , Davis BB , Wang ZH , Zhao SP , Wasti B , Liu ZL , Li N , Morisseau C , Chiamvimonvat N , Hammock BD |
| Ref : Int J Cardiol , 167 :1298 , 2013 |
| Abstract : Xu_2013_Int.J.Cardiol_167_1298 |
| ESTHER : Xu_2013_Int.J.Cardiol_167_1298 |
| PubMedSearch : Xu_2013_Int.J.Cardiol_167_1298 |
| PubMedID: 22525341 |
| Title : t-AUCB, an improved sEH inhibitor, suppresses human glioblastoma cell growth by activating NF-kappaB-p65 - Li_2012_J.Neurooncol_108_385 |
| Author(s) : Li J , Liu H , Xing B , Yu Y , Wang H , Chen G , Gu B , Zhang G , Wei D , Gu P , Li M , Hu W |
| Ref : J Neurooncol , 108 :385 , 2012 |
| Abstract : Li_2012_J.Neurooncol_108_385 |
| ESTHER : Li_2012_J.Neurooncol_108_385 |
| PubMedSearch : Li_2012_J.Neurooncol_108_385 |
| PubMedID: 22382785 |
| Title : The apoptosis-resistance in t-AUCB-treated glioblastoma cells depends on activation of Hsp27 - Li_2012_J.Neurooncol_110_187 |
| Author(s) : Li J , Hu W , Lan Q |
| Ref : J Neurooncol , 110 :187 , 2012 |
| Abstract : Li_2012_J.Neurooncol_110_187 |
| ESTHER : Li_2012_J.Neurooncol_110_187 |
| PubMedSearch : Li_2012_J.Neurooncol_110_187 |
| PubMedID: 22903412 |
| Title : [Soluble epoxide hydrolase inhibitor t-AUCB ameliorates ox-LDL induced conversion of macrophages into foam cells through activating the PPARgamma-ABCA1 pathway] - Zhao_2012_Zhonghua.Xin.Xue.Guan.Bing.Za.Zhi_40_248 |
| Author(s) : Zhao TT , Peng R , Shen L , Zhao X , Xu DY , Zhao SP |
| Ref : Zhonghua Xin Xue Guan Bing Za Zhi , 40 :248 , 2012 |
| Abstract : Zhao_2012_Zhonghua.Xin.Xue.Guan.Bing.Za.Zhi_40_248 |
| ESTHER : Zhao_2012_Zhonghua.Xin.Xue.Guan.Bing.Za.Zhi_40_248 |
| PubMedSearch : Zhao_2012_Zhonghua.Xin.Xue.Guan.Bing.Za.Zhi_40_248 |
| PubMedID: 22801272 |