Vinblastine
Vinblastine is a natural alkaloid isolated from the plant Vinca rosea Linn. Vinblastine is an antineoplastic agent. Vinblastine binds to tubulin and inhibits microtubule formation, resulting in disruption of mitotic spindle assembly and arrest of tumor cells in the M phase of the cell cycle. This agent may also interfere with amino acid, cyclic AMP, and glutathione metabolism; calmodulin-dependent Ca++ -transport ATPase activity; cellular respiration; and nucleic acid and lipid biosynthesis. It inhibits AADAC and CES2 but not CES1
General
Type : Alkaloid
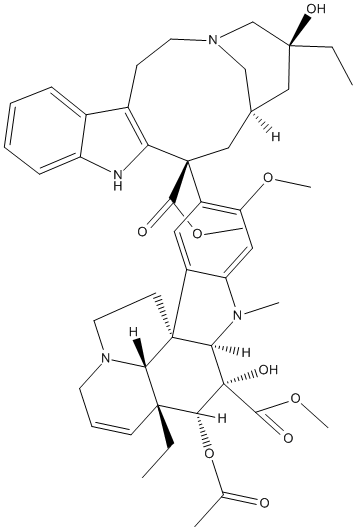
Chemical_Nomenclature : methyl (1R,9R,10S,11R,12R,19R)-11-acetyloxy-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-methoxycarbonyl-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10-hydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraene-10-carboxylate
Canonical SMILES : CCC1(CC2CC(C3=C(CCN(C2)C1)C4=CC=CC=C4N3)(C5=C(C=C6C(=C5)C78CCN9C7C(C=CC9)(C(C(C8N6C)(C(=O)OC)O)OC(=O)C)CC)OC)C(=O)OC)O
InChI : InChI=1S\/C46H58N4O9\/c1-8-42(54)23-28-24-45(40(52)57-6,36-30(15-19-49(25-28)26-42)29-13-10-11-14-33(29)47-36)32-21-31-34(22-35(32)56-5)48(4)38-44(31)17-20-50-18-12-16-43(9-2,37(44)50)39(59-27(3)51)46(38,55)41(53)58-7\/h10-14,16,21-22,28,37-39,47,54-55H,8-9,15,17-20,23-26H2,1-7H3\/t28-,37-,38+,39+,42-,43+,44+,45-,46-\/m0\/s1
InChIKey : JXLYSJRDGCGARV-CFWMRBGOSA-N
Other name(s) : Vinblastin,Vincaleucoblastin,Vinblastinum,Vincoblastine,Vinblastina,Velban
MW : 810.98
Formula : C46H58N4O9
CAS_number : 865-21-4
PubChem : 13342
UniChem : JXLYSJRDGCGARV-CFWMRBGOSA-N
IUPHAR :
Wikipedia : Vinblastine
Target
Families : Vinblastine ligand of proteins in family: Arylacetamide_deacetylase || Carb_B_Chordata
Stucture :
Protein : human-AADAC || human-CES2
References (2)
| Title : Role of human AADAC on hydrolysis of eslicarbazepine acetate and effects of AADAC genetic polymorphisms on hydrolase activity - Hirosawa_2021_Drug.Metab.Dispos__ |
| Author(s) : Hirosawa K , Fukami T , Tashiro K , Sakai Y , Kisui F , Nakano M , Nakajima M |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , : , 2021 |
| Abstract : Hirosawa_2021_Drug.Metab.Dispos__ |
| ESTHER : Hirosawa_2021_Drug.Metab.Dispos__ |
| PubMedSearch : Hirosawa_2021_Drug.Metab.Dispos__ |
| PubMedID: 33446525 |
| Gene_locus related to this paper: human-AADAC |
| Title : Screening of specific inhibitors for human carboxylesterases or arylacetamide deacetylase - Shimizu_2014_Drug.Metab.Dispos_42_1103 |
| Author(s) : Shimizu M , Fukami T , Nakajima M , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 42 :1103 , 2014 |
| Abstract : Shimizu_2014_Drug.Metab.Dispos_42_1103 |
| ESTHER : Shimizu_2014_Drug.Metab.Dispos_42_1103 |
| PubMedSearch : Shimizu_2014_Drug.Metab.Dispos_42_1103 |
| PubMedID: 24751575 |
| Gene_locus related to this paper: human-AADAC , human-CES1 , human-CES2 |