Talabostat
inhibitors of the serine peptidases DPP8 and DPP9 (DPP8/9) induce pro-caspase-1-dependent pyroptosis in monocytes and macrophages. inhibition of DPP8 and/or DPP9 has been shown to cause severe toxicity in preclinical species
General
Type : Peptide,Pyrrolidine,Boron compound
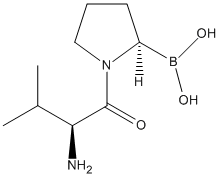
Chemical_Nomenclature : [(2R)-1-[(2S)-2-amino-3-methylbutanoyl]pyrrolidin-2-yl]boronic acid
Canonical SMILES : B(C1CCCN1C(=O)C(C(C)C)N)(O)O
InChI : InChI=1S\/C9H19BN2O3\/c1-6(2)8(11)9(13)12-5-3-4-7(12)10(14)15\/h6-8,14-15H,3-5,11H2,1-2H3\/t7-,8-\/m0\/s1
InChIKey : FKCMADOPPWWGNZ-YUMQZZPRSA-N
Other name(s) : Val-boro-pro,Val-boroPro,ValboroPro,UNII-KZ1O2SH88Z,Talabostat(PT100),VbP,PT-100,DB06182
MW : 214.07
Formula : C9H19BN2O3
CAS_number : 149682-77-9
PubChem : 6918572
UniChem : FKCMADOPPWWGNZ-YUMQZZPRSA-N
IUPHAR :
Wikipedia :
Target
Families : Talabostat ligand of proteins in family: DPP4N_Peptidase_S9
Stucture :
Protein : human-DPP8 || human-DPP9 || human-FAP
References (18)
| Title : DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation - Hollingsworth_2021_Nature_592_778 |
| Author(s) : Hollingsworth LR , Sharif H , Griswold AR , Fontana P , Mintseris J , Dagbay KB , Paulo JA , Gygi SP , Bachovchin DA , Wu H |
| Ref : Nature , 592 :778 , 2021 |
| Abstract : Hollingsworth_2021_Nature_592_778 |
| ESTHER : Hollingsworth_2021_Nature_592_778 |
| PubMedSearch : Hollingsworth_2021_Nature_592_778 |
| PubMedID: 33731932 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Dipeptidyl peptidase 9 sets a threshold for CARD8 inflammasome formation by sequestering its active C-terminal fragment - Sharif_2021_Immunity__ |
| Author(s) : Sharif H , Hollingsworth LR , Griswold AR , Hsiao JC , Wang Q , Bachovchin DA , Wu H |
| Ref : Immunity , : , 2021 |
| Abstract : Sharif_2021_Immunity__ |
| ESTHER : Sharif_2021_Immunity__ |
| PubMedSearch : Sharif_2021_Immunity__ |
| PubMedID: 34019797 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Structural mechanism of CARD8 regulation by DPP9 - Sharif_2021_Biorxiv__ |
| Author(s) : Sharif H , Hollingsworth LR , Griswold AR , Hsiao JC , Wang QH , Bachovchin DA , Wu H |
| Ref : Biorxiv , : , 2021 |
| Abstract : Sharif_2021_Biorxiv__ |
| ESTHER : Sharif_2021_Biorxiv__ |
| PubMedSearch : Sharif_2021_Biorxiv__ |
| PubMedID: |
| Gene_locus related to this paper: human-DPP9 |
| Title : DPP9 directly sequesters the NLRP1 C-terminus to repress inflammasome activation - Hollingsworth_2020_Biorxiv__ |
| Author(s) : Hollingsworth LR , Sharif H , Griswold AR , Fontana P , Mintseris J , Dagbay KB , Paulo JA , Gygi SP , Bachovchin DA , Wu |
| Ref : Biorxiv , : , 2020 |
| Abstract : Hollingsworth_2020_Biorxiv__ |
| ESTHER : Hollingsworth_2020_Biorxiv__ |
| PubMedSearch : Hollingsworth_2020_Biorxiv__ |
| PubMedID: |
| Gene_locus related to this paper: human-DPP9 |
| Title : The NLRP1 and CARD8 inflammasomes - Taabazuing_2020_Immunol.Rev_297_13 |
| Author(s) : Taabazuing CY , Griswold AR , Bachovchin DA |
| Ref : Immunol Rev , 297 :13 , 2020 |
| Abstract : Taabazuing_2020_Immunol.Rev_297_13 |
| ESTHER : Taabazuing_2020_Immunol.Rev_297_13 |
| PubMedSearch : Taabazuing_2020_Immunol.Rev_297_13 |
| PubMedID: 32558991 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Activation of the CARD8 Inflammasome Requires a Disordered Region - Chui_2020_Cell.Rep_33_108264 |
| Author(s) : Chui AJ , Griswold AR , Taabazuing CY , Orth EL , Gai K , Rao SD , Ball DP , Hsiao JC , Bachovchin DA |
| Ref : Cell Rep , 33 :108264 , 2020 |
| Abstract : Chui_2020_Cell.Rep_33_108264 |
| ESTHER : Chui_2020_Cell.Rep_33_108264 |
| PubMedSearch : Chui_2020_Cell.Rep_33_108264 |
| PubMedID: 33053349 |
| Gene_locus related to this paper: human-DPP8 , human-DPP9 |
| Title : DPP9's Enzymatic Activity and Not Its Binding to CARD8 Inhibits Inflammasome Activation - Griswold_2019_ACS.Chem.Biol_14_2424 |
| Author(s) : Griswold AR , Ball DP , Bhattacharjee A , Chui AJ , Rao SD , Taabazuing CY , Bachovchin DA |
| Ref : ACS Chemical Biology , 14 :2424 , 2019 |
| Abstract : Griswold_2019_ACS.Chem.Biol_14_2424 |
| ESTHER : Griswold_2019_ACS.Chem.Biol_14_2424 |
| PubMedSearch : Griswold_2019_ACS.Chem.Biol_14_2424 |
| PubMedID: 31525884 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding - Zhong_2018_J.Biol.Chem_293_18864 |
| Author(s) : Zhong FL , Robinson K , Teo DET , Tan KY , Lim C , Harapas CR , Yu CH , Xie WH , Sobota RM , Au VB , Hopkins R , D'Osualdo A , Reed JC , Connolly JE , Masters SL , Reversade B |
| Ref : Journal of Biological Chemistry , 293 :18864 , 2018 |
| Abstract : Zhong_2018_J.Biol.Chem_293_18864 |
| ESTHER : Zhong_2018_J.Biol.Chem_293_18864 |
| PubMedSearch : Zhong_2018_J.Biol.Chem_293_18864 |
| PubMedID: 30291141 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Inhibition of Dpp8\/9 Activates the Nlrp1b Inflammasome - Okondo_2018_Cell.Chem.Biol_25_262 |
| Author(s) : Okondo MC , Rao SD , Taabazuing CY , Chui AJ , Poplawski SE , Johnson DC , Bachovchin DA |
| Ref : Cell Chemical Biology , 25 :262 , 2018 |
| Abstract : Okondo_2018_Cell.Chem.Biol_25_262 |
| ESTHER : Okondo_2018_Cell.Chem.Biol_25_262 |
| PubMedSearch : Okondo_2018_Cell.Chem.Biol_25_262 |
| PubMedID: 29396289 |
| Gene_locus related to this paper: human-DPP8 , human-DPP9 |
| Title : Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages - Taabazuing_2017_Cell.Chem.Biol_24_507 |
| Author(s) : Taabazuing CY , Okondo MC , Bachovchin DA |
| Ref : Cell Chemical Biology , 24 :507 , 2017 |
| Abstract : Taabazuing_2017_Cell.Chem.Biol_24_507 |
| ESTHER : Taabazuing_2017_Cell.Chem.Biol_24_507 |
| PubMedSearch : Taabazuing_2017_Cell.Chem.Biol_24_507 |
| PubMedID: 28392147 |
| Gene_locus related to this paper: human-DPP8 , human-DPP9 |
| Title : DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis - Okondo_2017_Nat.Chem.Biol_13_46 |
| Author(s) : Okondo MC , Johnson DC , Sridharan R , Go EB , Chui AJ , Wang MS , Poplawski SE , Wu W , Liu Y , Lai JH , Sanford DG , Arciprete MO , Golub TR , Bachovchin WW , Bachovchin DA |
| Ref : Nat Chemical Biology , 13 :46 , 2017 |
| Abstract : Okondo_2017_Nat.Chem.Biol_13_46 |
| ESTHER : Okondo_2017_Nat.Chem.Biol_13_46 |
| PubMedSearch : Okondo_2017_Nat.Chem.Biol_13_46 |
| PubMedID: 27820798 |
| Gene_locus related to this paper: human-DPP8 , human-DPP9 |
| Title : Val-boroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors - Walsh_2013_PLoS.One_8_e58860 |
| Author(s) : Walsh MP , Duncan B , Larabee S , Krauss A , Davis JP , Cui Y , Kim SY , Guimond M , Bachovchin W , Fry TJ |
| Ref : PLoS ONE , 8 :e58860 , 2013 |
| Abstract : Walsh_2013_PLoS.One_8_e58860 |
| ESTHER : Walsh_2013_PLoS.One_8_e58860 |
| PubMedSearch : Walsh_2013_PLoS.One_8_e58860 |
| PubMedID: 23554941 |
| Title : 4-Substituted boro-proline dipeptides: synthesis, characterization, and dipeptidyl peptidase IV, 8, and 9 activities - Wu_2012_Bioorg.Med.Chem.Lett_22_5536 |
| Author(s) : Wu W , Liu Y , Milo LJ, Jr. , Shu Y , Zhao P , Li Y , Woznica I , Yu G , Sanford DG , Zhou Y , Poplawski SE , Connolly BA , Sudmeier JL , Bachovchin WW , Lai JH |
| Ref : Bioorganic & Medicinal Chemistry Lett , 22 :5536 , 2012 |
| Abstract : Wu_2012_Bioorg.Med.Chem.Lett_22_5536 |
| ESTHER : Wu_2012_Bioorg.Med.Chem.Lett_22_5536 |
| PubMedSearch : Wu_2012_Bioorg.Med.Chem.Lett_22_5536 |
| PubMedID: 22853995 |
| Title : Pro-soft Val-boroPro: a strategy for enhancing in vivo performance of boronic acid inhibitors of serine proteases - Poplawski_2011_J.Med.Chem_54_2022 |
| Author(s) : Poplawski SE , Lai JH , Sanford DG , Sudmeier JL , Wu W , Bachovchin WW |
| Ref : Journal of Medicinal Chemistry , 54 :2022 , 2011 |
| Abstract : Poplawski_2011_J.Med.Chem_54_2022 |
| ESTHER : Poplawski_2011_J.Med.Chem_54_2022 |
| PubMedSearch : Poplawski_2011_J.Med.Chem_54_2022 |
| PubMedID: 21388136 |
| Title : Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer - Eager_2009_Clin.Oncol.(R.Coll.Radiol)_21_464 |
| Author(s) : Eager RM , Cunningham CC , Senzer N , Richards DA , Raju RN , Jones B , Uprichard M , Nemunaitis J |
| Ref : Clin Oncol (R Coll Radiol) , 21 :464 , 2009 |
| Abstract : Eager_2009_Clin.Oncol.(R.Coll.Radiol)_21_464 |
| ESTHER : Eager_2009_Clin.Oncol.(R.Coll.Radiol)_21_464 |
| PubMedSearch : Eager_2009_Clin.Oncol.(R.Coll.Radiol)_21_464 |
| PubMedID: 19501491 |
| Title : Dipeptide boronic acid inhibitors of dipeptidyl peptidase IV: determinants of potency and in vivo efficacy and safety - Connolly_2008_J.Med.Chem_51_6005 |
| Author(s) : Connolly BA , Sanford DG , Chiluwal AK , Healey SE , Peters DE , Dimare MT , Wu W , Liu Y , Maw H , Zhou Y , Li Y , Jin Z , Sudmeier JL , Lai JH , Bachovchin WW |
| Ref : Journal of Medicinal Chemistry , 51 :6005 , 2008 |
| Abstract : Connolly_2008_J.Med.Chem_51_6005 |
| ESTHER : Connolly_2008_J.Med.Chem_51_6005 |
| PubMedSearch : Connolly_2008_J.Med.Chem_51_6005 |
| PubMedID: 18783201 |
| Title : Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer - Narra_2007_Cancer.Biol.Ther_6_1691 |
| Author(s) : Narra K , Mullins SR , Lee HO , Strzemkowski-Brun B , Magalong K , Christiansen VJ , McKee PA , Egleston B , Cohen SJ , Weiner LM , Meropol NJ , Cheng JD |
| Ref : Cancer Biol Ther , 6 :1691 , 2007 |
| Abstract : Narra_2007_Cancer.Biol.Ther_6_1691 |
| ESTHER : Narra_2007_Cancer.Biol.Ther_6_1691 |
| PubMedSearch : Narra_2007_Cancer.Biol.Ther_6_1691 |
| PubMedID: 18032930 |
| Title : Effect of deoxycoformycin and Val-boroPro on the associated catalytic activities of lymphocyte CD26 and ecto-adenosine deaminase - Jeanfavre_1996_Biochem.Pharmacol_52_1757 |
| Author(s) : Jeanfavre DD , Woska JR, Jr. , Pargellis CA , Kennedy CA , Prendergast J , Stearns C , Reilly PL , Barton RW , Bormann BJ |
| Ref : Biochemical Pharmacology , 52 :1757 , 1996 |
| Abstract : Jeanfavre_1996_Biochem.Pharmacol_52_1757 |
| ESTHER : Jeanfavre_1996_Biochem.Pharmacol_52_1757 |
| PubMedSearch : Jeanfavre_1996_Biochem.Pharmacol_52_1757 |
| PubMedID: 8986139 |