TBTFA
Uncharged transition state analogue of TMTFA
General
Type : Halo ketone,Transition state analogue,Trifluoro,tert-Butyl
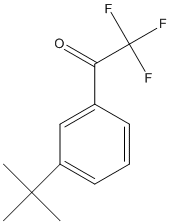
Chemical_Nomenclature : 1-(3-tert-butylphenyl)-2,2,2-trifluoroethanone
Canonical SMILES : CC(C)(C)C1=CC=CC(=C1)C(=O)C(F)(F)F
InChI : InChI=1S\/C12H13F3O\/c1-11(2,3)9-6-4-5-8(7-9)10(16)12(13,14)15\/h4-7H,1-3H3
InChIKey : XAZQUPBUJONHAF-UHFFFAOYSA-N
Other name(s) : m-(t-butyl)trifluoroacetophenone,m-(t-butyl)-2,2,2-Trifluoro-1,1-dihydroxyethylbenzene,CHEMBL89354,3'-tert-Butyl-2,2,2-trifluoroacetophenone,155628-03-8,SCHEMBL7651441,M-tertbutyl trifluoroacetophenone,1-(3-Tert-Butylphenyl)-2,2,2-Trifluoroethanone,m-(Tert-butyl) trifluoroacetophenone,ZINC1540476,TFKo,MolPort-035-781-534,BDBM50281126
MW : 230.23
Formula : C12H13F3O
CAS_number :
PubChem : 9859434
UniChem : XAZQUPBUJONHAF-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (7)
| Title : Drug and pro-drug substrates and pseudo-substrates of human butyrylcholinesterase - Masson_2023_Biochem.Pharmacol_218_115910 |
| Author(s) : Masson P , Shaihutdinova Z , Lockridge O |
| Ref : Biochemical Pharmacology , 218 :115910 , 2023 |
| Abstract : Masson_2023_Biochem.Pharmacol_218_115910 |
| ESTHER : Masson_2023_Biochem.Pharmacol_218_115910 |
| PubMedSearch : Masson_2023_Biochem.Pharmacol_218_115910 |
| PubMedID: 37972875 |
| Title : Protective effects of m-(tert-butyl) trifluoroacetophenone, a transition state analogue of acetylcholine, against paraoxon toxicity and memory impairments - Zueva_2021_Chem.Biol.Interact_345_109558 |
| Author(s) : Zueva IV , Lenina OA , Kayumova RM , Petrov KA , Masson P |
| Ref : Chemico-Biological Interactions , 345 :109558 , 2021 |
| Abstract : Zueva_2021_Chem.Biol.Interact_345_109558 |
| ESTHER : Zueva_2021_Chem.Biol.Interact_345_109558 |
| PubMedSearch : Zueva_2021_Chem.Biol.Interact_345_109558 |
| PubMedID: 34147486 |
| Title : 1-(3-Tert-Butylphenyl)-2,2,2-Trifluoroethanone as a Potent Transition-State Analogue Slow-Binding Inhibitor of Human Acetylcholinesterase: Kinetic, MD and QM\/MM Studies - Zueva_2020_Biomolecules_10_1608 |
| Author(s) : Zueva IV , Lushchekina SV , Pottie IR , Darvesh S , Masson P |
| Ref : Biomolecules , 10 : , 2020 |
| Abstract : Zueva_2020_Biomolecules_10_1608 |
| ESTHER : Zueva_2020_Biomolecules_10_1608 |
| PubMedSearch : Zueva_2020_Biomolecules_10_1608 |
| PubMedID: 33260981 |
| Title : Development of acetophenone ligands as potential neuroimaging agents for cholinesterases - Jollymore-Hughes_2016_Bioorg.Med.Chem_24_5270 |
| Author(s) : Jollymore-Hughes CT , Pottie IR , Martin E , Rosenberry TL , Darvesh S |
| Ref : Bioorganic & Medicinal Chemistry , 24 :5270 , 2016 |
| Abstract : Jollymore-Hughes_2016_Bioorg.Med.Chem_24_5270 |
| ESTHER : Jollymore-Hughes_2016_Bioorg.Med.Chem_24_5270 |
| PubMedSearch : Jollymore-Hughes_2016_Bioorg.Med.Chem_24_5270 |
| PubMedID: 27637382 |
| Title : Theoretical and experimental investigations of electrostatic effects on acetylcholinesterase catalysis and inhibition - Malany_1999_Chem.Biol.Interact_119-120_99 |
| Author(s) : Malany S , Baker NA , Verweyst M , Medhekar R , Quinn DM , Velan B , Kronman C , Shafferman A |
| Ref : Chemico-Biological Interactions , 119-120 :99 , 1999 |
| Abstract : Malany_1999_Chem.Biol.Interact_119-120_99 |
| ESTHER : Malany_1999_Chem.Biol.Interact_119-120_99 |
| PubMedSearch : Malany_1999_Chem.Biol.Interact_119-120_99 |
| PubMedID: 10421443 |
| Title : Allosteric control of acetylcholinesterase catalysis by fasciculin - Radic_1995_J.Biol.Chem_270_20391 |
| Author(s) : Radic Z , Quinn DM , Vellom DC , Camp S , Taylor P |
| Ref : Journal of Biological Chemistry , 270 :20391 , 1995 |
| Abstract : Radic_1995_J.Biol.Chem_270_20391 |
| ESTHER : Radic_1995_J.Biol.Chem_270_20391 |
| PubMedSearch : Radic_1995_J.Biol.Chem_270_20391 |
| PubMedID: 7657613 |
| Title : Molecular recognition in acetylcholinesterase catalysis: free-energy correlations for substrate turnover and inhibition by trifluoro ketone transition-state analogs - Nair_1994_Biochemistry_33_8566 |
| Author(s) : Nair HK , Seravalli J , Arbuckle T , Quinn DM |
| Ref : Biochemistry , 33 :8566 , 1994 |
| Abstract : Nair_1994_Biochemistry_33_8566 |
| ESTHER : Nair_1994_Biochemistry_33_8566 |
| PubMedSearch : Nair_1994_Biochemistry_33_8566 |
| PubMedID: 8031791 |