Sitagliptin
Sitagliptin is a pyrazine-derived dipeptidyl-peptidase IV inhibitor and hypoglycemic agent that is used in conjunction with diet and exercise to improve glycemic control in patients with type 2 diabetes mellitus. The effect of this medication leads to glucose dependent increases in insulin and decreases in glucagon to improve control of blood sugar. Modulates the levels of the incretin hormones glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide (GIP). CAS_Number 2243737-33-7 cetagliptin phosphate
General
Type : Drug,Piperidine,Gliptin,Triazol,Trifluoro
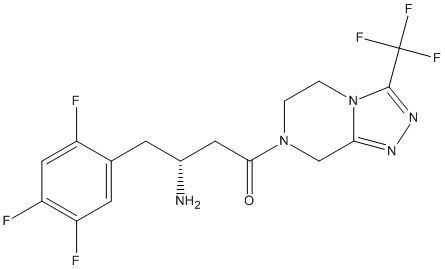
Chemical_Nomenclature : (3R)-3-amino-1-[3-(trifluoromethyl)-6,8-dihydro-5H-[1,2,4]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one.
Canonical SMILES : C1CN2C(=NN=C2C(F)(F)F)CN1C(=O)CC(CC3=CC(=C(C=C3F)F)F)N
InChI : InChI=1S\/C16H15F6N5O\/c17-10-6-12(19)11(18)4-8(10)3-9(23)5-14(28)26-1-2-27-13(7-26)24-25-15(27)16(20,21)22\/h4,6,9H,1-3,5,7,23H2\/t9-\/m1\/s1
InChIKey : MFFMDFFZMYYVKS-SECBINFHSA-N
Other name(s) : Januvia,Xelevia,MK-0431,MK0431,Sitagliptan,715,DB01261,Cetagliptin,CHEBI:40237,CHEMBL1422,SCHEMBL17783
MW : 407.31
Formula : C16H15F6N5O
CAS_number : 486460-32-6 || 2243737-33-7
PubChem : 4369359
UniChem : MFFMDFFZMYYVKS-SECBINFHSA-N
IUPHAR : 6286
Wikipedia : Sitagliptin
Target
Families : Sitagliptin ligand of proteins in family: DPP4N_Peptidase_S9
Stucture :
Protein : human-DPP4 || bactn-BT4193
References (97)
| Title : Safety, tolerability, pharmacokinetics and pharmacokinetic-pharmacodynamic modeling of cetagliptin in patients with type 2 diabetes mellitus - Zhou_2024_Front.Endocrinol.(Lausanne)_15_1359407 |
| Author(s) : Zhou C , Zhou S , Wang J , Xie L , Lv Z , Zhao Y , Wang L , Luo H , Xie D , Shao F |
| Ref : Front Endocrinol (Lausanne) , 15 :1359407 , 2024 |
| Abstract : Zhou_2024_Front.Endocrinol.(Lausanne)_15_1359407 |
| ESTHER : Zhou_2024_Front.Endocrinol.(Lausanne)_15_1359407 |
| PubMedSearch : Zhou_2024_Front.Endocrinol.(Lausanne)_15_1359407 |
| PubMedID: 38529396 |
| Title : Efficacy and safety of cetagliptin as monotherapy in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled phase 3 trial - Ji_2023_Diabetes.Obes.Metab_25_3671 |
| Author(s) : Ji L , Lu J , Gao L , Ying C , Sun J , Han J , Zhao W , Gao Y , Wang K , Zheng X , Xie D , Ding J , Zhao J , Yu Q , Wang T |
| Ref : Diabetes Obes Metab , 25 :3671 , 2023 |
| Abstract : Ji_2023_Diabetes.Obes.Metab_25_3671 |
| ESTHER : Ji_2023_Diabetes.Obes.Metab_25_3671 |
| PubMedSearch : Ji_2023_Diabetes.Obes.Metab_25_3671 |
| PubMedID: 37661308 |
| Title : A randomized, double-blind, placebo controlled, phase 3 trial to evaluate the efficacy and safety of cetagliptin added to ongoing metformin therapy in patients with uncontrolled type 2 diabetes with metformin monotherapy - Ji_2023_Diabetes.Obes.Metab_25_3788 |
| Author(s) : Ji L , Lu J , Gao L , Yan X , Li J , Cheng Z , Zhang L , Tian J , Li P , Bai J , Xie D , Zhao J , Ding J , Yu Q , Wang T |
| Ref : Diabetes Obes Metab , 25 :3788 , 2023 |
| Abstract : Ji_2023_Diabetes.Obes.Metab_25_3788 |
| ESTHER : Ji_2023_Diabetes.Obes.Metab_25_3788 |
| PubMedSearch : Ji_2023_Diabetes.Obes.Metab_25_3788 |
| PubMedID: 37724698 |
| Title : Sitagliptin Extends Lifespan of Caenorhabditis elegans by Inhibiting IIS and Activating DR-like Signaling Pathways - Ye_2023_Gerontology__ |
| Author(s) : Ye Q , Li Y , Wang C , Zheng J , Qiao J , Yang J , Wan QL |
| Ref : Gerontology , : , 2023 |
| Abstract : Ye_2023_Gerontology__ |
| ESTHER : Ye_2023_Gerontology__ |
| PubMedSearch : Ye_2023_Gerontology__ |
| PubMedID: 37952525 |
| Title : Mechanism of molecular interaction of sitagliptin with human DPP(4) enzyme - New Insights - Gonzatti_2023_Adv.Med.Sci_68_402 |
| Author(s) : Gonzatti MB , Junior JEM , Rocha AJ , de Oliveira JS , Evangelista AJJ , Fonseca FMP , Ceccatto VM , de Oliveira AC , da Cruz Freire JE |
| Ref : Adv Med Sci , 68 :402 , 2023 |
| Abstract : Gonzatti_2023_Adv.Med.Sci_68_402 |
| ESTHER : Gonzatti_2023_Adv.Med.Sci_68_402 |
| PubMedSearch : Gonzatti_2023_Adv.Med.Sci_68_402 |
| PubMedID: 37837799 |
| Title : Antidiabetic drug sitagliptin blocks cyclophosphamide cerebral neurotoxicity by activating Nrf2 and suppressing redox cycle imbalance, inflammatory iNOS\/NO\/NF-B response and caspase-3\/Bax activation in rats - Famurewa_2023_Int.Immunopharmacol_116_109816 |
| Author(s) : Famurewa AC , Asogwa NT , Ezea SC |
| Ref : Int Immunopharmacol , 116 :109816 , 2023 |
| Abstract : Famurewa_2023_Int.Immunopharmacol_116_109816 |
| ESTHER : Famurewa_2023_Int.Immunopharmacol_116_109816 |
| PubMedSearch : Famurewa_2023_Int.Immunopharmacol_116_109816 |
| PubMedID: 36774854 |
| Title : Targeting DPP4-RBD interactions by sitagliptin and linagliptin delivers a potential host-directed therapy against pan-SARS-CoV-2 infections - Mani_2023_Int.J.Biol.Macromol_245_125444 |
| Author(s) : Mani S , Kaur A , Jakhar K , Kumari G , Sonar S , Kumar A , Das S , Kumar S , Kumar V , Kundu R , Pandey AK , Singh UP , Majumdar T |
| Ref : Int J Biol Macromol , 245 :125444 , 2023 |
| Abstract : Mani_2023_Int.J.Biol.Macromol_245_125444 |
| ESTHER : Mani_2023_Int.J.Biol.Macromol_245_125444 |
| PubMedSearch : Mani_2023_Int.J.Biol.Macromol_245_125444 |
| PubMedID: 37385308 |
| Gene_locus related to this paper: human-DPP4 |
| Title : Sitagliptin attenuates neuronal apoptosis via inhibiting the endoplasmic reticulum stress after acute spinal cord injury - Tang_2023_Hum.Exp.Toxicol_42_9603271231168761 |
| Author(s) : Tang C , Xu T , Dai M , Zhong X , Shen G , Liu L |
| Ref : Hum Exp Toxicol , 42 :9603271231168761 , 2023 |
| Abstract : Tang_2023_Hum.Exp.Toxicol_42_9603271231168761 |
| ESTHER : Tang_2023_Hum.Exp.Toxicol_42_9603271231168761 |
| PubMedSearch : Tang_2023_Hum.Exp.Toxicol_42_9603271231168761 |
| PubMedID: 36977492 |
| Title : The metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [(14)C] cetagliptin in healthy volunteers - Lu_2022_Xenobiotica_52_38 |
| Author(s) : Lu J , Bian Y , Zhang H , Tang D , Tian X , Zhou X , Xu Z , Xiong Y , Gu Z , Yu Z , Wang T , Ding J , Yu Q |
| Ref : Xenobiotica , 52 :38 , 2022 |
| Abstract : Lu_2022_Xenobiotica_52_38 |
| ESTHER : Lu_2022_Xenobiotica_52_38 |
| PubMedSearch : Lu_2022_Xenobiotica_52_38 |
| PubMedID: 34743655 |
| Title : Disposition study of the novel dipeptidyl peptidase 4 inhibitor cetagliptin in rats - Lu_2022_Xenobiotica_52_468 |
| Author(s) : Lu J , Hao Y , Zhang F , Pan H , Ding J , Yu Q , Wang T |
| Ref : Xenobiotica , 52 :468 , 2022 |
| Abstract : Lu_2022_Xenobiotica_52_468 |
| ESTHER : Lu_2022_Xenobiotica_52_468 |
| PubMedSearch : Lu_2022_Xenobiotica_52_468 |
| PubMedID: 35708192 |
| Title : A double-blind, randomized, placebo and positive-controlled study in healthy volunteers to evaluate pharmacokinetic and pharmacodynamic properties of multiple oral doses of cetagliptin - Lu_2022_Br.J.Clin.Pharmacol_88_2946 |
| Author(s) : Lu J , Wang L , Zhou S , Zhou C , Xie L , Chen J , Tang D , Tian X , Xie D , Ding J , Wang T , Yu Q , Shao F |
| Ref : British Journal of Clinical Pharmacology , 88 :2946 , 2022 |
| Abstract : Lu_2022_Br.J.Clin.Pharmacol_88_2946 |
| ESTHER : Lu_2022_Br.J.Clin.Pharmacol_88_2946 |
| PubMedSearch : Lu_2022_Br.J.Clin.Pharmacol_88_2946 |
| PubMedID: 34965609 |
| Title : Sitagliptin Alleviates Radiation-Induced Intestinal Injury by Activating NRF2-Antioxidant Axis, Mitigating NLRP3 Inf--lammasome Activation, and Reversing Gut Microbiota Disorder - Huang_2022_Oxid.Med.Cell.Longev_2022_2586305 |
| Author(s) : Huang S , Huang Y , Lin W , Wang L , Yang Y , Li P , Xiao L , Chen Y , Chu Q , Yuan X |
| Ref : Oxid Med Cell Longev , 2022 :2586305 , 2022 |
| Abstract : Huang_2022_Oxid.Med.Cell.Longev_2022_2586305 |
| ESTHER : Huang_2022_Oxid.Med.Cell.Longev_2022_2586305 |
| PubMedSearch : Huang_2022_Oxid.Med.Cell.Longev_2022_2586305 |
| PubMedID: 35620578 |
| Title : Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Exhibits Antioxidant Mechanism for Abrogation of Cyclophosphamide-Induced Cardiac Damage and Oxidative Hepatorenal Toxicity in Rats - Famurewa_2022_Drug.Res.(Stuttg)__ |
| Author(s) : Famurewa AC , Aja PM , Medewase JO , Abi I , Ogbonna OC , Ofor CC , Nwonuma CO , Asogwa NT , Erejuwa OO |
| Ref : Drug Res (Stuttg) , : , 2022 |
| Abstract : Famurewa_2022_Drug.Res.(Stuttg)__ |
| ESTHER : Famurewa_2022_Drug.Res.(Stuttg)__ |
| PubMedSearch : Famurewa_2022_Drug.Res.(Stuttg)__ |
| PubMedID: 35772725 |
| Title : Evaluation of the anti-diabetic drug sitagliptin as a novel attenuate to SARS-CoV-2 evidence-based in silico: molecular docking and molecular dynamics - da Cruz Freire_2022_3.Biotech_12_344 |
| Author(s) : da Cruz Freire JE , Jnior JEM , Pinheiro DP , da Cruz Paiva Lima GE , do Amaral CL , Veras VR , Madeira MP , Freire EBL , Ozrio RG , Fernandes VO , Montenegro A , Montenegro RC , Colares JKB , Jnior RMM |
| Ref : 3 Biotech , 12 :344 , 2022 |
| Abstract : da Cruz Freire_2022_3.Biotech_12_344 |
| ESTHER : da Cruz Freire_2022_3.Biotech_12_344 |
| PubMedSearch : da Cruz Freire_2022_3.Biotech_12_344 |
| PubMedID: 36382241 |
| Title : A Rare Case Report of Sitagliptin-Induced Angioedema - Sharma_2022_Cureus_14_e30077 |
| Author(s) : Sharma NR , Sharma B , Lamichhane S , Pokhrel M , Shrestha P |
| Ref : Cureus , 14 :e30077 , 2022 |
| Abstract : Sharma_2022_Cureus_14_e30077 |
| ESTHER : Sharma_2022_Cureus_14_e30077 |
| PubMedSearch : Sharma_2022_Cureus_14_e30077 |
| PubMedID: 36381841 |
| Title : Development and validation of an ultrasensitive LC-MS\/MS method for the quantification of cetagliptin in human plasma and its application in a microdose clinical trial - Bai_2021_Biomed.Chromatogr_35_e4994 |
| Author(s) : Bai H , Lu J , Cheng X , Liu L , Zhang W , Wei Y , Wang Y , Liu J , Ding J , Yu Q , Zhang Y , Chen G , Fan Y , Wang X |
| Ref : Biomedical Chromatography , 35 :e4994 , 2021 |
| Abstract : Bai_2021_Biomed.Chromatogr_35_e4994 |
| ESTHER : Bai_2021_Biomed.Chromatogr_35_e4994 |
| PubMedSearch : Bai_2021_Biomed.Chromatogr_35_e4994 |
| PubMedID: 32986878 |
| Title : In vitro study of the drug-drug interaction potential of cetagliptin and clinical study of pharmacokinetic interaction of cetagliptin and metformin in healthy volunteers - Lu_2021_Xenobiotica_51_1122 |
| Author(s) : Lu J , Tian X , Tang D , Zhou X , Xu Z , Ding J , Wang T , Yu Q |
| Ref : Xenobiotica , 51 :1122 , 2021 |
| Abstract : Lu_2021_Xenobiotica_51_1122 |
| ESTHER : Lu_2021_Xenobiotica_51_1122 |
| PubMedSearch : Lu_2021_Xenobiotica_51_1122 |
| PubMedID: 34329567 |
| Title : First-in-Human, Single-Ascending Dose and Food Effect Studies to Assess the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Cetagliptin, a Dipeptidyl Peptidase-4 Inhibitor for the Treatment of Type 2 Diabetes Mellitus - Wang_2021_Clin.Drug.Investig_41_999 |
| Author(s) : Wang L , Lu J , Zhou S , Zhao Y , Xie L , Zhou C , Chen J , Ding S , Xie D , Ding J , Yu Q , Shen H , Hao G , Shao F |
| Ref : Clin Drug Investig , 41 :999 , 2021 |
| Abstract : Wang_2021_Clin.Drug.Investig_41_999 |
| ESTHER : Wang_2021_Clin.Drug.Investig_41_999 |
| PubMedSearch : Wang_2021_Clin.Drug.Investig_41_999 |
| PubMedID: 34655432 |
| Title : Sitagliptin: a potential drug for the treatment of COVID-19? - Bardaweel_2021_Acta.Pharm_71_175 |
| Author(s) : Bardaweel SK , Hajjo R , Sabbah DA |
| Ref : Acta Pharm , 71 :175 , 2021 |
| Abstract : Bardaweel_2021_Acta.Pharm_71_175 |
| ESTHER : Bardaweel_2021_Acta.Pharm_71_175 |
| PubMedSearch : Bardaweel_2021_Acta.Pharm_71_175 |
| PubMedID: 33151168 |
| Title : Promoter Engineering-mediated Tuning of Esterase and Transaminase Expression for the Chemoenzymatic Synthesis of Sitagliptin Phosphate at the kilogram-scale - Khobragade_2021_Biotechnol.Bioeng__ |
| Author(s) : Khobragade TP , Yu S , Jung H , Patil MD , Sarak S , Pagar AD , Jeon H , Lee S , Giri P , Kim GH , Cho SS , Park SH , Park HJ , Kang HM , Lee SR , Lee MS , Kim JH , Choi IS , Yun H |
| Ref : Biotechnol Bioeng , : , 2021 |
| Abstract : Khobragade_2021_Biotechnol.Bioeng__ |
| ESTHER : Khobragade_2021_Biotechnol.Bioeng__ |
| PubMedSearch : Khobragade_2021_Biotechnol.Bioeng__ |
| PubMedID: 33990942 |
| Title : Combined use of GABA and sitagliptin promotes human beta-cell proliferation and reduces apoptosis - Liu_2021_J.Endocrinol_248_133 |
| Author(s) : Liu W , Lau HK , Son DO , Jin T , Yang Y , Zhang Z , Li Y , Prud'homme GJ , Wang Q |
| Ref : J Endocrinol , 248 :133 , 2021 |
| Abstract : Liu_2021_J.Endocrinol_248_133 |
| ESTHER : Liu_2021_J.Endocrinol_248_133 |
| PubMedSearch : Liu_2021_J.Endocrinol_248_133 |
| PubMedID: 33258801 |
| Title : Sitagliptin Modulates the Response of Ovarian Cancer Cells to Chemotherapeutic Agents - Kosowska_2020_Int.J.Mol.Sci_21_ |
| Author(s) : Kosowska A , Garczorz W , Klych-Ratuszny A , Aghdam MRF , Kimsa-Furdzik M , Simka-Lampa K , Francuz T |
| Ref : Int J Mol Sci , 21 : , 2020 |
| Abstract : Kosowska_2020_Int.J.Mol.Sci_21_ |
| ESTHER : Kosowska_2020_Int.J.Mol.Sci_21_ |
| PubMedSearch : Kosowska_2020_Int.J.Mol.Sci_21_ |
| PubMedID: 33256016 |
| Title : Higher-Dose Sitagliptin and the Risk of Congestive Heart Failure in Older Adults with CKD - Muanda_2020_Clin.J.Am.Soc.Nephrol_15_1728 |
| Author(s) : Muanda FT , Weir MA , Bathini L , Clemens KK , Perkovic V , Sood MM , McArthur E , Sontrop JM , Kim RB , Garg AX |
| Ref : Clin J Am Soc Nephrol , 15 :1728 , 2020 |
| Abstract : Muanda_2020_Clin.J.Am.Soc.Nephrol_15_1728 |
| ESTHER : Muanda_2020_Clin.J.Am.Soc.Nephrol_15_1728 |
| PubMedSearch : Muanda_2020_Clin.J.Am.Soc.Nephrol_15_1728 |
| PubMedID: 33239410 |
| Title : Saxagliptin but Not Sitagliptin Inhibits CaMKII and PKC via DPP9 Inhibition in Cardiomyocytes - Koyani_2018_Front.Physiol_9_1622 |
| Author(s) : Koyani CN , Trummer C , Shrestha N , Scheruebel S , Bourgeois B , Plastira I , Kickmaier S , Sourij H , Rainer PP , Madl T , Sattler W , Pelzmann B , Malle E , von Lewinski D |
| Ref : Front Physiol , 9 :1622 , 2018 |
| Abstract : Koyani_2018_Front.Physiol_9_1622 |
| ESTHER : Koyani_2018_Front.Physiol_9_1622 |
| PubMedSearch : Koyani_2018_Front.Physiol_9_1622 |
| PubMedID: 30487758 |
| Gene_locus related to this paper: human-DPP9 |
| Title : Effect of Anagliptin and Sitagliptin in Type 2 Diabetic Patients with Dyslipidemia and Cardiovascular Risk: What Is the Real REASON Behind It? - |
| Author(s) : Jonnalagadda VG , Char HP , Ankana PK |
| Ref : Cardiovasc Drugs Ther , 32 :311 , 2018 |
| PubMedID: 29516206 |
| Title : A comparative study of the binding properties, dipeptidyl peptidase-4 (DPP-4) inhibitory activity and glucose-lowering efficacy of the DPP-4 inhibitors alogliptin, linagliptin, saxagliptin, sitagliptin and vildagliptin in mice - Berger_2018_Endocrinol.Diabetes.Metab_1_e00002 |
| Author(s) : Berger JP , SinhaRoy R , Pocai A , Kelly TM , Scapin G , Gao YD , Pryor KAD , Wu JK , Eiermann GJ , Xu SS , Zhang X , Tatosian DA , Weber AE , Thornberry NA , Carr RD |
| Ref : Endocrinol Diabetes Metab , 1 :e00002 , 2018 |
| Abstract : Berger_2018_Endocrinol.Diabetes.Metab_1_e00002 |
| ESTHER : Berger_2018_Endocrinol.Diabetes.Metab_1_e00002 |
| PubMedSearch : Berger_2018_Endocrinol.Diabetes.Metab_1_e00002 |
| PubMedID: 30815539 |
| Gene_locus related to this paper: human-DPP4 |
| Title : Effect of Anagliptin and Sitagliptin on Low-Density Lipoprotein Cholesterol in Type 2 Diabetic Patients with Dyslipidemia and Cardiovascular Risk: Rationale and Study Design of the REASON Trial - Ueda_2018_Cardiovasc.Drugs.Ther_32_73 |
| Author(s) : Ueda S , Shimabukuro M , Arasaki O , Node K , Nomiyama T , Morimoto T |
| Ref : Cardiovasc Drugs Ther , 32 :73 , 2018 |
| Abstract : Ueda_2018_Cardiovasc.Drugs.Ther_32_73 |
| ESTHER : Ueda_2018_Cardiovasc.Drugs.Ther_32_73 |
| PubMedSearch : Ueda_2018_Cardiovasc.Drugs.Ther_32_73 |
| PubMedID: 29435776 |
| Title : Efficacy and safety of adding evogliptin versus sitagliptin for metformin-treated patients with type 2 diabetes: A 24-week randomized, controlled trial with open label extension - Hong_2017_Diabetes.Obes.Metab_19_654 |
| Author(s) : Hong SM , Park CY , Hwang DM , Han KA , Lee CB , Chung CH , Yoon KH , Mok JO , Park KS , Park SW |
| Ref : Diabetes Obes Metab , 19 :654 , 2017 |
| Abstract : Hong_2017_Diabetes.Obes.Metab_19_654 |
| ESTHER : Hong_2017_Diabetes.Obes.Metab_19_654 |
| PubMedSearch : Hong_2017_Diabetes.Obes.Metab_19_654 |
| PubMedID: 28058750 |
| Title : Association of Sitagliptin with cardiovascular outcome in diabetic patients: a nationwide cohort study - Yang_2016_Acta.Diabetol_53_461 |
| Author(s) : Yang TY , Liaw YP , Huang JY , Chang HR , Chang KW , Ueng KC |
| Ref : Acta Diabetologia , 53 :461 , 2016 |
| Abstract : Yang_2016_Acta.Diabetol_53_461 |
| ESTHER : Yang_2016_Acta.Diabetol_53_461 |
| PubMedSearch : Yang_2016_Acta.Diabetol_53_461 |
| PubMedID: 26687195 |
| Title : Sitagliptin, a dipeptidyl peptidase-4 inhibitor, improves recognition memory, oxidative stress and hippocampal neurogenesis and upregulates key genes involved in cognitive decline - Gault_2015_Diabetes.Obes.Metab_17_403 |
| Author(s) : Gault VA , Lennox R , Flatt PR |
| Ref : Diabetes Obes Metab , 17 :403 , 2015 |
| Abstract : Gault_2015_Diabetes.Obes.Metab_17_403 |
| ESTHER : Gault_2015_Diabetes.Obes.Metab_17_403 |
| PubMedSearch : Gault_2015_Diabetes.Obes.Metab_17_403 |
| PubMedID: 25580570 |
| Title : Anagliptin and sitagliptin as add-ons to metformin for patients with type 2 diabetes: a 24-week, multicentre, randomized, double-blind, active-controlled, phase III clinical trial with a 28-week extension - Jin_2015_Diabetes.Obes.Metab_17_511 |
| Author(s) : Jin SM , Park SW , Yoon KH , Min KW , Song KH , Park KS , Park JY , Park IB , Chung CH , Baik SH , Choi SH , Lee HW , Lee IK , Kim DM , Lee MK |
| Ref : Diabetes Obes Metab , 17 :511 , 2015 |
| Abstract : Jin_2015_Diabetes.Obes.Metab_17_511 |
| ESTHER : Jin_2015_Diabetes.Obes.Metab_17_511 |
| PubMedSearch : Jin_2015_Diabetes.Obes.Metab_17_511 |
| PubMedID: 25523633 |
| Title : Retrospective and Prospective Human Intravenous and Oral Pharmacokinetic Projection of Dipeptidyl peptidase-IV Inhibitors Using Simple Allometric Principles - Case Studies of ABT-279, ABT-341, Alogliptin, Carmegliptin, Sitagliptin and Vildagliptin - Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| Author(s) : Gilibili RR , Bhamidipati RK , Mullangi R , Srinivas NR |
| Ref : J Pharm Pharm Sci , 18 :434 , 2015 |
| Abstract : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| ESTHER : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| PubMedSearch : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| PubMedID: 26517136 |
| Title : Therapeutic effects of the dipeptidyl peptidase-IV inhibitor, sitagliptin, on non-alcoholic steatohepatitis in FLS-ob\/ob male mice - Onoyama_2015_Mol.Med.Rep_12_6895 |
| Author(s) : Onoyama T , Koda M , Okamoto T , Kishina M , Matono T , Sugihara T , Murawaki Y |
| Ref : Mol Med Rep , 12 :6895 , 2015 |
| Abstract : Onoyama_2015_Mol.Med.Rep_12_6895 |
| ESTHER : Onoyama_2015_Mol.Med.Rep_12_6895 |
| PubMedSearch : Onoyama_2015_Mol.Med.Rep_12_6895 |
| PubMedID: 26397061 |
| Title : An Inhibitory Antibody against Dipeptidyl Peptidase IV Improves Glucose Tolerance in Vivo - Tang_2013_J.Biol.Chem_288_1307 |
| Author(s) : Tang J , Majeti J , Sudom A , Xiong Y , Lu M , Liu Q , Higbee J , Zhang Y , Wang Y , Wang W , Cao P , Xia Z , Johnstone S , Min X , Yang X , Shao H , Yu T , Sharkov N , Walker N , Tu H , Shen W , Wang Z |
| Ref : Journal of Biological Chemistry , 288 :1307 , 2013 |
| Abstract : Tang_2013_J.Biol.Chem_288_1307 |
| ESTHER : Tang_2013_J.Biol.Chem_288_1307 |
| PubMedSearch : Tang_2013_J.Biol.Chem_288_1307 |
| PubMedID: 23184939 |
| Gene_locus related to this paper: ratno-dpp4 |
| Title : Sitagliptin, a dipeptidyl peptidase-4 inhibitor, does not affect the pharmacokinetics of ethinyl estradiol or norethindrone in healthy female subjects - Migoya_2011_J.Clin.Pharmacol_51_1319 |
| Author(s) : Migoya E , Larson P , Bergman A , Miller J , Johnson-Levonas AO , Lasseter KC , Wagner JA |
| Ref : Journal of Clinical Pharmacology , 51 :1319 , 2011 |
| Abstract : Migoya_2011_J.Clin.Pharmacol_51_1319 |
| ESTHER : Migoya_2011_J.Clin.Pharmacol_51_1319 |
| PubMedSearch : Migoya_2011_J.Clin.Pharmacol_51_1319 |
| PubMedID: 21209231 |
| Title : Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice - Dobrian_2011_Am.J.Physiol.Endocrinol.Metab_300_E410 |
| Author(s) : Dobrian AD , Ma Q , Lindsay JW , Leone KA , Ma K , Coben J , Galkina EV , Nadler JL |
| Ref : American Journal of Physiology Endocrinol Metab , 300 :E410 , 2011 |
| Abstract : Dobrian_2011_Am.J.Physiol.Endocrinol.Metab_300_E410 |
| ESTHER : Dobrian_2011_Am.J.Physiol.Endocrinol.Metab_300_E410 |
| PubMedSearch : Dobrian_2011_Am.J.Physiol.Endocrinol.Metab_300_E410 |
| PubMedID: 21081706 |
| Title : Evaluation of pharmacokinetic parameters and dipeptidyl peptidase-4 inhibition following single doses of sitagliptin in healthy, young Japanese males - Herman_2011_Br.J.Clin.Pharmacol_71_429 |
| Author(s) : Herman GA , Mistry GC , Yi B , Bergman AJ , Wang AQ , Zeng W , Chen L , Snyder K , Ruckle JL , Larson PJ , Davies MJ , Langdon RB , Gottesdiener KM , Wagner JA |
| Ref : British Journal of Clinical Pharmacology , 71 :429 , 2011 |
| Abstract : Herman_2011_Br.J.Clin.Pharmacol_71_429 |
| ESTHER : Herman_2011_Br.J.Clin.Pharmacol_71_429 |
| PubMedSearch : Herman_2011_Br.J.Clin.Pharmacol_71_429 |
| PubMedID: 21284702 |
| Title : Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and pioglitazone on glycemic control and measures of beta-cell function in patients with type 2 diabetes - Yoon_2011_Int.J.Clin.Pract_65_154 |
| Author(s) : Yoon KH , Shockey GR , Teng R , Golm GT , Thakkar PR , Meehan AG , Williams-Herman DE , Kaufman KD , Amatruda JM , Steinberg H |
| Ref : Int J Clin Pract , 65 :154 , 2011 |
| Abstract : Yoon_2011_Int.J.Clin.Pract_65_154 |
| ESTHER : Yoon_2011_Int.J.Clin.Pract_65_154 |
| PubMedSearch : Yoon_2011_Int.J.Clin.Pract_65_154 |
| PubMedID: 21235696 |
| Title : Sitagliptin: review of preclinical and clinical data regarding incidence of pancreatitis - Engel_2010_Int.J.Clin.Pract_64_984 |
| Author(s) : Engel SS , Williams-Herman DE , Golm GT , Clay RJ , Machotka SV , Kaufman KD , Goldstein BJ |
| Ref : Int J Clin Pract , 64 :984 , 2010 |
| Abstract : Engel_2010_Int.J.Clin.Pract_64_984 |
| ESTHER : Engel_2010_Int.J.Clin.Pract_64_984 |
| PubMedSearch : Engel_2010_Int.J.Clin.Pract_64_984 |
| PubMedID: 20412332 |
| Title : The effect of DPP-4 inhibition with sitagliptin on incretin secretion and on fasting and postprandial glucose turnover in subjects with impaired fasting glucose - Bock_2010_Clin.Endocrinol.(Oxf)_73_189 |
| Author(s) : Bock G , Dalla Man C , Micheletto F , Basu R , Giesler PD , Laugen J , Deacon CF , Holst JJ , Toffolo G , Cobelli C , Rizza RA , Vella A |
| Ref : Clinical Endocrinology (Oxf) , 73 :189 , 2010 |
| Abstract : Bock_2010_Clin.Endocrinol.(Oxf)_73_189 |
| ESTHER : Bock_2010_Clin.Endocrinol.(Oxf)_73_189 |
| PubMedSearch : Bock_2010_Clin.Endocrinol.(Oxf)_73_189 |
| PubMedID: 20039889 |
| Title : The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha - Fadini_2010_Diabetes.Care_33_1607 |
| Author(s) : Fadini GP , Boscaro E , Albiero M , Menegazzo L , Frison V , de Kreutzenberg S , Agostini C , Tiengo A , Avogaro A |
| Ref : Diabetes Care , 33 :1607 , 2010 |
| Abstract : Fadini_2010_Diabetes.Care_33_1607 |
| ESTHER : Fadini_2010_Diabetes.Care_33_1607 |
| PubMedSearch : Fadini_2010_Diabetes.Care_33_1607 |
| PubMedID: 20357375 |
| Title : Dose-ranging efficacy of sitagliptin, a dipeptidyl peptidase-4 inhibitor, in Japanese patients with type 2 diabetes mellitus - Iwamoto_2010_Endocr.J_57_383 |
| Author(s) : Iwamoto Y , Taniguchi T , Nonaka K , Okamoto T , Okuyama K , Arjona Ferreira JC , Amatruda J |
| Ref : Endocrine Journal , 57 :383 , 2010 |
| Abstract : Iwamoto_2010_Endocr.J_57_383 |
| ESTHER : Iwamoto_2010_Endocr.J_57_383 |
| PubMedSearch : Iwamoto_2010_Endocr.J_57_383 |
| PubMedID: 20332588 |
| Title : Effects of miglitol, sitagliptin or their combination on plasma glucose, insulin and incretin levels in non-diabetic men - Aoki_2010_Endocr.J_57_667 |
| Author(s) : Aoki K , Masuda K , Miyazaki T , Togashi Y , Terauchi Y |
| Ref : Endocrine Journal , 57 :667 , 2010 |
| Abstract : Aoki_2010_Endocr.J_57_667 |
| ESTHER : Aoki_2010_Endocr.J_57_667 |
| PubMedSearch : Aoki_2010_Endocr.J_57_667 |
| PubMedID: 20519806 |
| Title : Sitagliptin: a review of its use in the management of type 2 diabetes mellitus - Dhillon_2010_Drugs_70_489 |
| Author(s) : Dhillon S |
| Ref : Drugs , 70 :489 , 2010 |
| Abstract : Dhillon_2010_Drugs_70_489 |
| ESTHER : Dhillon_2010_Drugs_70_489 |
| PubMedSearch : Dhillon_2010_Drugs_70_489 |
| PubMedID: 20205490 |
| Title : Sitagliptin (MK0431) inhibition of dipeptidyl peptidase IV decreases nonobese diabetic mouse CD4+ T-cell migration through incretin-dependent and -independent pathways - Kim_2010_Diabetes_59_1739 |
| Author(s) : Kim SJ , Nian C , McIntosh CH |
| Ref : Diabetes , 59 :1739 , 2010 |
| Abstract : Kim_2010_Diabetes_59_1739 |
| ESTHER : Kim_2010_Diabetes_59_1739 |
| PubMedSearch : Kim_2010_Diabetes_59_1739 |
| PubMedID: 20368408 |
| Title : Safety and tolerability of sitagliptin in clinical studies: a pooled analysis of data from 10,246 patients with type 2 diabetes - Williams-Herman_2010_BMC.Endocr.Disord_10_7 |
| Author(s) : Williams-Herman D , Engel SS , Round E , Johnson J , Golm GT , Guo H , Musser BJ , Davies MJ , Kaufman KD , Goldstein BJ |
| Ref : BMC Endocr Disord , 10 :7 , 2010 |
| Abstract : Williams-Herman_2010_BMC.Endocr.Disord_10_7 |
| ESTHER : Williams-Herman_2010_BMC.Endocr.Disord_10_7 |
| PubMedSearch : Williams-Herman_2010_BMC.Endocr.Disord_10_7 |
| PubMedID: 20412573 |
| Title : Nature of action of Sitagliptin, the dipeptidyl peptidase-IV inhibitor in diabetic animals - Davis_2010_Indian.J.Pharmacol_42_229 |
| Author(s) : Davis JA , Singh S , Sethi S , Roy S , Mittra S , Rayasam G , Bansal V , Sattigeri J , Ray A |
| Ref : Indian J Pharmacol , 42 :229 , 2010 |
| Abstract : Davis_2010_Indian.J.Pharmacol_42_229 |
| ESTHER : Davis_2010_Indian.J.Pharmacol_42_229 |
| PubMedSearch : Davis_2010_Indian.J.Pharmacol_42_229 |
| PubMedID: 20927248 |
| Title : Effects of vildagliptin twice daily vs. sitagliptin once daily on 24-hour acute glucose fluctuations - Marfella_2010_J.Diabetes.Complications_24_79 |
| Author(s) : Marfella R , Barbieri M , Grella R , Rizzo MR , Nicoletti GF , Paolisso G |
| Ref : J Diabetes Complications , 24 :79 , 2010 |
| Abstract : Marfella_2010_J.Diabetes.Complications_24_79 |
| ESTHER : Marfella_2010_J.Diabetes.Complications_24_79 |
| PubMedSearch : Marfella_2010_J.Diabetes.Complications_24_79 |
| PubMedID: 19261490 |
| Title : DPP4 inhibitors: from sitagliptin monotherapy to the new alogliptin-pioglitazone combination therapy - Argyrakopoulou_2009_Adv.Ther_26_272 |
| Author(s) : Argyrakopoulou G , Doupis J |
| Ref : Adv Ther , 26 :272 , 2009 |
| Abstract : Argyrakopoulou_2009_Adv.Ther_26_272 |
| ESTHER : Argyrakopoulou_2009_Adv.Ther_26_272 |
| PubMedSearch : Argyrakopoulou_2009_Adv.Ther_26_272 |
| PubMedID: 19259628 |
| Title : Sitagliptin 100 mg daily effect on DPP-4 inhibition and compound-specific glycemic improvement - Alba_2009_Curr.Med.Res.Opin_25_2507 |
| Author(s) : Alba M , Sheng D , Guan Y , Williams-Herman D , Larson P , Sachs JR , Thornberry N , Herman G , Kaufman KD , Goldstein BJ |
| Ref : Curr Med Res Opin , 25 :2507 , 2009 |
| Abstract : Alba_2009_Curr.Med.Res.Opin_25_2507 |
| ESTHER : Alba_2009_Curr.Med.Res.Opin_25_2507 |
| PubMedSearch : Alba_2009_Curr.Med.Res.Opin_25_2507 |
| PubMedID: 19691426 |
| Title : Enhancement of hematopoietic stem cell engraftment by inhibition of CXCL12 proteolysis with sitagliptin, an oral dipeptidyl-peptidase IV inhibitor: a report in a case of delayed graft failure - |
| Author(s) : Focosi D , Kast RE , Metelli MR , Benedetti E , Galimberti S , Papineschi F , Petrini M |
| Ref : Leuk Res , 33 :178 , 2009 |
| PubMedID: 18513794 |
| Title : Clinical results of treating type 2 diabetic patients with sitagliptin, vildagliptin or saxagliptin--diabetes control and potential adverse events - Ahren_2009_Best.Pract.Res.Clin.Endocrinol.Metab_23_487 |
| Author(s) : Ahren B |
| Ref : Best Pract Res Clinical Endocrinology Metab , 23 :487 , 2009 |
| Abstract : Ahren_2009_Best.Pract.Res.Clin.Endocrinol.Metab_23_487 |
| ESTHER : Ahren_2009_Best.Pract.Res.Clin.Endocrinol.Metab_23_487 |
| PubMedSearch : Ahren_2009_Best.Pract.Res.Clin.Endocrinol.Metab_23_487 |
| PubMedID: 19748066 |
| Title : Impact of sitagliptin on markers of beta-cell function: a meta-analysis - Riche_2009_Am.J.Med.Sci_337_321 |
| Author(s) : Riche DM , East HE , Riche KD |
| Ref : American Journal of Medicine Sci , 337 :321 , 2009 |
| Abstract : Riche_2009_Am.J.Med.Sci_337_321 |
| ESTHER : Riche_2009_Am.J.Med.Sci_337_321 |
| PubMedSearch : Riche_2009_Am.J.Med.Sci_337_321 |
| PubMedID: 19322069 |
| Title : Multiple doses of sitagliptin, a selective DPP-4 inhibitor, do not meaningfully alter pharmacokinetics and pharmacodynamics of warfarin - Wright_2009_J.Clin.Pharmacol_49_1157 |
| Author(s) : Wright DH , Herman GA , Maes A , Liu Q , Johnson-Levonas AO , Wagner JA |
| Ref : Journal of Clinical Pharmacology , 49 :1157 , 2009 |
| Abstract : Wright_2009_J.Clin.Pharmacol_49_1157 |
| ESTHER : Wright_2009_J.Clin.Pharmacol_49_1157 |
| PubMedSearch : Wright_2009_J.Clin.Pharmacol_49_1157 |
| PubMedID: 19783710 |
| Title : Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes - Mu_2009_Eur.J.Pharmacol_623_148 |
| Author(s) : Mu J , Petrov A , Eiermann GJ , Woods J , Zhou YP , Li Z , Zycband E , Feng Y , Zhu L , Roy RS , Howard AD , Li C , Thornberry NA , Zhang BB |
| Ref : European Journal of Pharmacology , 623 :148 , 2009 |
| Abstract : Mu_2009_Eur.J.Pharmacol_623_148 |
| ESTHER : Mu_2009_Eur.J.Pharmacol_623_148 |
| PubMedSearch : Mu_2009_Eur.J.Pharmacol_623_148 |
| PubMedID: 19765579 |
| Title : [New drugs\; exenatide and sitagliptin] - van Bronswijk_2008_Ned.Tijdschr.Geneeskd_152_876 |
| Author(s) : van Bronswijk H , Dubois EA , Pijl H , Cohen AF |
| Ref : Ned Tijdschr Geneeskd , 152 :876 , 2008 |
| Abstract : van Bronswijk_2008_Ned.Tijdschr.Geneeskd_152_876 |
| ESTHER : van Bronswijk_2008_Ned.Tijdschr.Geneeskd_152_876 |
| PubMedSearch : van Bronswijk_2008_Ned.Tijdschr.Geneeskd_152_876 |
| PubMedID: 18512528 |
| Title : Sitagliptin: a novel agent for the management of type 2 diabetes mellitus - Pham_2008_Am.J.Health.Syst.Pharm_65_521 |
| Author(s) : Pham DQ , Nogid A , Plakogiannis R |
| Ref : Am J Health Syst Pharm , 65 :521 , 2008 |
| Abstract : Pham_2008_Am.J.Health.Syst.Pharm_65_521 |
| ESTHER : Pham_2008_Am.J.Health.Syst.Pharm_65_521 |
| PubMedSearch : Pham_2008_Am.J.Health.Syst.Pharm_65_521 |
| PubMedID: 18319497 |
| Title : Safety and tolerability of sitagliptin in patients with type 2 diabetes: a pooled analysis - Williams-Herman_2008_BMC.Endocr.Disord_8_14 |
| Author(s) : Williams-Herman D , Round E , Swern AS , Musser B , Davies MJ , Stein PP , Kaufman KD , Amatruda JM |
| Ref : BMC Endocr Disord , 8 :14 , 2008 |
| Abstract : Williams-Herman_2008_BMC.Endocr.Disord_8_14 |
| ESTHER : Williams-Herman_2008_BMC.Endocr.Disord_8_14 |
| PubMedSearch : Williams-Herman_2008_BMC.Endocr.Disord_8_14 |
| PubMedID: 18954434 |
| Title : New combination treatments in the management of diabetes: focus on sitagliptin-metformin - Green_2008_Vasc.Health.Risk.Manag_4_743 |
| Author(s) : Green J , Feinglos M |
| Ref : Vasc Health Risk Manag , 4 :743 , 2008 |
| Abstract : Green_2008_Vasc.Health.Risk.Manag_4_743 |
| ESTHER : Green_2008_Vasc.Health.Risk.Manag_4_743 |
| PubMedSearch : Green_2008_Vasc.Health.Risk.Manag_4_743 |
| PubMedID: 19065992 |
| Title : Sitagliptin augments sympathetic enhancement of the renovascular effects of angiotensin II in genetic hypertension - Jackson_2008_Hypertension_51_1637 |
| Author(s) : Jackson EK , Mi Z |
| Ref : Hypertension , 51 :1637 , 2008 |
| Abstract : Jackson_2008_Hypertension_51_1637 |
| ESTHER : Jackson_2008_Hypertension_51_1637 |
| PubMedSearch : Jackson_2008_Hypertension_51_1637 |
| PubMedID: 18443229 |
| Title : New treatments for type 2 diabetes mellitus: combined therapy with sitagliptin - Gagliardino_2008_Expert.Opin.Pharmacother_9_1495 |
| Author(s) : Gagliardino JJ , Santoro S , Arellano S , Di Girolamo G |
| Ref : Expert Opin Pharmacother , 9 :1495 , 2008 |
| Abstract : Gagliardino_2008_Expert.Opin.Pharmacother_9_1495 |
| ESTHER : Gagliardino_2008_Expert.Opin.Pharmacother_9_1495 |
| PubMedSearch : Gagliardino_2008_Expert.Opin.Pharmacother_9_1495 |
| PubMedID: 18518780 |
| Title : Inhibition of dipeptidyl peptidase IV with sitagliptin (MK0431) prolongs islet graft survival in streptozotocin-induced diabetic mice - Kim_2008_Diabetes_57_1331 |
| Author(s) : Kim SJ , Nian C , Doudet DJ , McIntosh CH |
| Ref : Diabetes , 57 :1331 , 2008 |
| Abstract : Kim_2008_Diabetes_57_1331 |
| ESTHER : Kim_2008_Diabetes_57_1331 |
| PubMedSearch : Kim_2008_Diabetes_57_1331 |
| PubMedID: 18299314 |
| Title : Sitagliptin, an dipeptidyl peptidase-4 inhibitor, does not alter the pharmacokinetics of the sulphonylurea, glyburide, in healthy subjects - Mistry_2008_Br.J.Clin.Pharmacol_66_36 |
| Author(s) : Mistry GC , Bergman AJ , Zheng W , Hreniuk D , Zinny MA , Gottesdiener KM , Wagner JA , Herman GA , Ruddy M |
| Ref : British Journal of Clinical Pharmacology , 66 :36 , 2008 |
| Abstract : Mistry_2008_Br.J.Clin.Pharmacol_66_36 |
| ESTHER : Mistry_2008_Br.J.Clin.Pharmacol_66_36 |
| PubMedSearch : Mistry_2008_Br.J.Clin.Pharmacol_66_36 |
| PubMedID: 18503607 |
| Title : Effect of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on beta-cell function in patients with type 2 diabetes: a model-based approach - Xu_2008_Diabetes.Obes.Metab_10_1212 |
| Author(s) : Xu L , Man CD , Charbonnel B , Meninger G , Davies MJ , Williams-Herman D , Cobelli C , Stein PP |
| Ref : Diabetes Obes Metab , 10 :1212 , 2008 |
| Abstract : Xu_2008_Diabetes.Obes.Metab_10_1212 |
| ESTHER : Xu_2008_Diabetes.Obes.Metab_10_1212 |
| PubMedSearch : Xu_2008_Diabetes.Obes.Metab_10_1212 |
| PubMedID: 18476982 |
| Title : Effect of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on blood pressure in nondiabetic patients with mild to moderate hypertension - Mistry_2008_J.Clin.Pharmacol_48_592 |
| Author(s) : Mistry GC , Maes AL , Lasseter KC , Davies MJ , Gottesdiener KM , Wagner JA , Herman GA |
| Ref : Journal of Clinical Pharmacology , 48 :592 , 2008 |
| Abstract : Mistry_2008_J.Clin.Pharmacol_48_592 |
| ESTHER : Mistry_2008_J.Clin.Pharmacol_48_592 |
| PubMedSearch : Mistry_2008_J.Clin.Pharmacol_48_592 |
| PubMedID: 18353996 |
| Title : [Incretin strategy in the treatment of type 2 diabetes mellitus--the DPP-IV inhibitor sitagliptin] - Perusicova_2007_Vnitr.Lek_53_1109 |
| Author(s) : Perusicova J |
| Ref : Vnitr Lek , 53 :1109 , 2007 |
| Abstract : Perusicova_2007_Vnitr.Lek_53_1109 |
| ESTHER : Perusicova_2007_Vnitr.Lek_53_1109 |
| PubMedSearch : Perusicova_2007_Vnitr.Lek_53_1109 |
| PubMedID: 18072437 |
| Title : Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor - |
| Author(s) : Bergman AJ , Cote J , Yi B , Marbury T , Swan SK , Smith W , Gottesdiener K , Wagner J , Herman GA |
| Ref : Diabetes Care , 30 :1862 , 2007 |
| PubMedID: 17468348 |
| Title : Sitagliptin - |
| Author(s) : Drucker D , Easley C , Kirkpatrick P |
| Ref : Nat Rev Drug Discov , 6 :109 , 2007 |
| PubMedID: 17342863 |
| Title : Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes - Goldstein_2007_Diabetes.Care_30_1979 |
| Author(s) : Goldstein BJ , Feinglos MN , Lunceford JK , Johnson J , Williams-Herman DE |
| Ref : Diabetes Care , 30 :1979 , 2007 |
| Abstract : Goldstein_2007_Diabetes.Care_30_1979 |
| ESTHER : Goldstein_2007_Diabetes.Care_30_1979 |
| PubMedSearch : Goldstein_2007_Diabetes.Care_30_1979 |
| PubMedID: 17485570 |
| Title : Sitagliptin: a novel drug for the treatment of type 2 diabetes - Choy_2007_Cardiol.Rev_15_264 |
| Author(s) : Choy M , Lam S |
| Ref : Cardiol Rev , 15 :264 , 2007 |
| Abstract : Choy_2007_Cardiol.Rev_15_264 |
| ESTHER : Choy_2007_Cardiol.Rev_15_264 |
| PubMedSearch : Choy_2007_Cardiol.Rev_15_264 |
| PubMedID: 17700385 |
| Title : Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein - Chu_2007_J.Pharmacol.Exp.Ther_321_673 |
| Author(s) : Chu XY , Bleasby K , Yabut J , Cai X , Chan GH , Hafey MJ , Xu S , Bergman AJ , Braun MP , Dean DC , Evers R |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 321 :673 , 2007 |
| Abstract : Chu_2007_J.Pharmacol.Exp.Ther_321_673 |
| ESTHER : Chu_2007_J.Pharmacol.Exp.Ther_321_673 |
| PubMedSearch : Chu_2007_J.Pharmacol.Exp.Ther_321_673 |
| PubMedID: 17314201 |
| Title : Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin - Hermansen_2007_Diabetes.Obes.Metab_9_733 |
| Author(s) : Hermansen K , Kipnes M , Luo E , Fanurik D , Khatami H , Stein P |
| Ref : Diabetes Obes Metab , 9 :733 , 2007 |
| Abstract : Hermansen_2007_Diabetes.Obes.Metab_9_733 |
| ESTHER : Hermansen_2007_Diabetes.Obes.Metab_9_733 |
| PubMedSearch : Hermansen_2007_Diabetes.Obes.Metab_9_733 |
| PubMedID: 17593236 |
| Title : Disposition of the dipeptidyl peptidase 4 inhibitor sitagliptin in rats and dogs - Beconi_2007_Drug.Metab.Dispos_35_525 |
| Author(s) : Beconi MG , Reed JR , Teffera Y , Xia YQ , Kochansky CJ , Liu DQ , Xu S , Elmore CS , Ciccotto S , Hora DF , Stearns RA , Vincent SH |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 35 :525 , 2007 |
| Abstract : Beconi_2007_Drug.Metab.Dispos_35_525 |
| ESTHER : Beconi_2007_Drug.Metab.Dispos_35_525 |
| PubMedSearch : Beconi_2007_Drug.Metab.Dispos_35_525 |
| PubMedID: 17220241 |
| Title : Dipeptidyl peptidase 4 inhibition with sitagliptin: a new therapy for type 2 diabetes - Deacon_2007_Expert.Opin.Investig.Drugs_16_533 |
| Author(s) : Deacon CF |
| Ref : Expert Opin Investig Drugs , 16 :533 , 2007 |
| Abstract : Deacon_2007_Expert.Opin.Investig.Drugs_16_533 |
| ESTHER : Deacon_2007_Expert.Opin.Investig.Drugs_16_533 |
| PubMedSearch : Deacon_2007_Expert.Opin.Investig.Drugs_16_533 |
| PubMedID: 17371200 |
| Title : Sitagliptin: Profile of a novel DPP-4 inhibitor for the treatment of type 2 diabetes - Gallwitz_2007_Drugs.Today.(Barc)_43_13 |
| Author(s) : Gallwitz B |
| Ref : Drugs Today (Barc) , 43 :13 , 2007 |
| Abstract : Gallwitz_2007_Drugs.Today.(Barc)_43_13 |
| ESTHER : Gallwitz_2007_Drugs.Today.(Barc)_43_13 |
| PubMedSearch : Gallwitz_2007_Drugs.Today.(Barc)_43_13 |
| PubMedID: 17315049 |
| Title : Triazolopiperazine-amides as dipeptidyl peptidase IV inhibitors: close analogs of JANUVIA (sitagliptin phosphate) - Kim_2007_Bioorg.Med.Chem.Lett_17_3373 |
| Author(s) : Kim D , Kowalchick JE , Edmondson SD , Mastracchio A , Xu J , Eiermann GJ , Leiting B , Wu JK , Pryor KD , Patel RA , He H , Lyons KA , Thornberry NA , Weber AE |
| Ref : Bioorganic & Medicinal Chemistry Lett , 17 :3373 , 2007 |
| Abstract : Kim_2007_Bioorg.Med.Chem.Lett_17_3373 |
| ESTHER : Kim_2007_Bioorg.Med.Chem.Lett_17_3373 |
| PubMedSearch : Kim_2007_Bioorg.Med.Chem.Lett_17_3373 |
| PubMedID: 17434732 |
| Title : Sitagliptin with metformin: profile of a combination for the treatment of type 2 diabetes - Gallwitz_2007_Drugs.Today.(Barc)_43_681 |
| Author(s) : Gallwitz B |
| Ref : Drugs Today (Barc) , 43 :681 , 2007 |
| Abstract : Gallwitz_2007_Drugs.Today.(Barc)_43_681 |
| ESTHER : Gallwitz_2007_Drugs.Today.(Barc)_43_681 |
| PubMedSearch : Gallwitz_2007_Drugs.Today.(Barc)_43_681 |
| PubMedID: 17987221 |
| Title : Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: focus on sitagliptin - Herman_2007_Clin.Pharmacol.Ther_81_761 |
| Author(s) : Herman GA , Stein PP , Thornberry NA , Wagner JA |
| Ref : Clinical Pharmacology & Therapeutics , 81 :761 , 2007 |
| Abstract : Herman_2007_Clin.Pharmacol.Ther_81_761 |
| ESTHER : Herman_2007_Clin.Pharmacol.Ther_81_761 |
| PubMedSearch : Herman_2007_Clin.Pharmacol.Ther_81_761 |
| PubMedID: 17392725 |
| Title : Sitagliptin: profile of a novel DPP-4 inhibitor for the treatment of type 2 diabetes (update) - Gallwitz_2007_Drugs.Today.(Barc)_43_801 |
| Author(s) : Gallwitz B |
| Ref : Drugs Today (Barc) , 43 :801 , 2007 |
| Abstract : Gallwitz_2007_Drugs.Today.(Barc)_43_801 |
| ESTHER : Gallwitz_2007_Drugs.Today.(Barc)_43_801 |
| PubMedSearch : Gallwitz_2007_Drugs.Today.(Barc)_43_801 |
| PubMedID: 18174966 |
| Title : Rational design of a novel, potent, and orally bioavailable cyclohexylamine DPP-4 inhibitor by application of molecular modeling and X-ray crystallography of sitagliptin - Biftu_2007_Bioorg.Med.Chem.Lett_17_3384 |
| Author(s) : Biftu T , Scapin G , Singh S , Feng D , Becker JW , Eiermann G , He H , Lyons K , Patel S , Petrov A , Sinha-Roy R , Zhang B , Wu J , Zhang X , Doss GA , Thornberry NA , Weber AE |
| Ref : Bioorganic & Medicinal Chemistry Lett , 17 :3384 , 2007 |
| Abstract : Biftu_2007_Bioorg.Med.Chem.Lett_17_3384 |
| ESTHER : Biftu_2007_Bioorg.Med.Chem.Lett_17_3384 |
| PubMedSearch : Biftu_2007_Bioorg.Med.Chem.Lett_17_3384 |
| PubMedID: 17433672 |
| Gene_locus related to this paper: human-DPP4 |
| Title : Multiple-dose administration of sitagliptin, a dipeptidyl peptidase-4 inhibitor, does not alter the single-dose pharmacokinetics of rosiglitazone in healthy subjects - Mistry_2007_J.Clin.Pharmacol_47_159 |
| Author(s) : Mistry GC , Bergman AJ , Luo WL , Cilissen C , Haazen W , Davies MJ , Gottesdiener KM , Wagner JA , Herman GA |
| Ref : Journal of Clinical Pharmacology , 47 :159 , 2007 |
| Abstract : Mistry_2007_J.Clin.Pharmacol_47_159 |
| ESTHER : Mistry_2007_J.Clin.Pharmacol_47_159 |
| PubMedSearch : Mistry_2007_J.Clin.Pharmacol_47_159 |
| PubMedID: 17244766 |
| Title : Effect of a single cyclosporine dose on the single-dose pharmacokinetics of sitagliptin (MK-0431), a dipeptidyl peptidase-4 inhibitor, in healthy male subjects - Krishna_2007_J.Clin.Pharmacol_47_165 |
| Author(s) : Krishna R , Bergman A , Larson P , Cote J , Lasseter K , Dilzer S , Wang A , Zeng W , Chen L , Wagner J , Herman G |
| Ref : Journal of Clinical Pharmacology , 47 :165 , 2007 |
| Abstract : Krishna_2007_J.Clin.Pharmacol_47_165 |
| ESTHER : Krishna_2007_J.Clin.Pharmacol_47_165 |
| PubMedSearch : Krishna_2007_J.Clin.Pharmacol_47_165 |
| PubMedID: 17244767 |
| Title : Characterization of two cyclic metabolites of sitagliptin - Liu_2007_Drug.Metab.Dispos_35_521 |
| Author(s) : Liu DQ , Arison BH , Stearns RA , Kim D , Vincent SH |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 35 :521 , 2007 |
| Abstract : Liu_2007_Drug.Metab.Dispos_35_521 |
| ESTHER : Liu_2007_Drug.Metab.Dispos_35_521 |
| PubMedSearch : Liu_2007_Drug.Metab.Dispos_35_521 |
| PubMedID: 17220240 |
| Title : Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes - Scott_2007_Int.J.Clin.Pract_61_171 |
| Author(s) : Scott R , Wu M , Sanchez M , Stein P |
| Ref : Int J Clin Pract , 61 :171 , 2007 |
| Abstract : Scott_2007_Int.J.Clin.Pract_61_171 |
| ESTHER : Scott_2007_Int.J.Clin.Pract_61_171 |
| PubMedSearch : Scott_2007_Int.J.Clin.Pract_61_171 |
| PubMedID: 17156104 |
| Title : Sitagliptin - Lyseng-Williamson_2007_Drugs_67_587 |
| Author(s) : Lyseng-Williamson KA |
| Ref : Drugs , 67 :587 , 2007 |
| Abstract : Lyseng-Williamson_2007_Drugs_67_587 |
| ESTHER : Lyseng-Williamson_2007_Drugs_67_587 |
| PubMedSearch : Lyseng-Williamson_2007_Drugs_67_587 |
| PubMedID: 17352516 |
| Title : Sitagliptin phosphate: a DPP-4 inhibitor for the treatment of type 2 diabetes mellitus - Zerilli_2007_Clin.Ther_29_2614 |
| Author(s) : Zerilli T , Pyon EY |
| Ref : Clin Ther , 29 :2614 , 2007 |
| Abstract : Zerilli_2007_Clin.Ther_29_2614 |
| ESTHER : Zerilli_2007_Clin.Ther_29_2614 |
| PubMedSearch : Zerilli_2007_Clin.Ther_29_2614 |
| PubMedID: 18201579 |
| Title : Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes - Thornberry_2007_Curr.Top.Med.Chem_7_557 |
| Author(s) : Thornberry NA , Weber AE |
| Ref : Curr Top Med Chem , 7 :557 , 2007 |
| Abstract : Thornberry_2007_Curr.Top.Med.Chem_7_557 |
| ESTHER : Thornberry_2007_Curr.Top.Med.Chem_7_557 |
| PubMedSearch : Thornberry_2007_Curr.Top.Med.Chem_7_557 |
| PubMedID: 17352677 |
| Title : Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects - Herman_2006_J.Clin.Pharmacol_46_876 |
| Author(s) : Herman GA , Bergman A , Liu F , Stevens C , Wang AQ , Zeng W , Chen L , Snyder K , Hilliard D , Tanen M , Tanaka W , Meehan AG , Lasseter K , Dilzer S , Blum R , Wagner JA |
| Ref : Journal of Clinical Pharmacology , 46 :876 , 2006 |
| Abstract : Herman_2006_J.Clin.Pharmacol_46_876 |
| ESTHER : Herman_2006_J.Clin.Pharmacol_46_876 |
| PubMedSearch : Herman_2006_J.Clin.Pharmacol_46_876 |
| PubMedID: 16855072 |
| Title : Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes - Herman_2006_J.Clin.Endocrinol.Metab_91_4612 |
| Author(s) : Herman GA , Bergman A , Stevens C , Kotey P , Yi B , Zhao P , Dietrich B , Golor G , Schrodter A , Keymeulen B , Lasseter KC , Kipnes MS , Snyder K , Hilliard D , Tanen M , Cilissen C , De Smet M , De Lepeleire I , Van Dyck K , Wang AQ , Zeng W , Davies MJ , Tanaka W , Holst JJ , Deacon CF , Gottesdiener KM , Wagner JA |
| Ref : J Clinical Endocrinology Metab , 91 :4612 , 2006 |
| Abstract : Herman_2006_J.Clin.Endocrinol.Metab_91_4612 |
| ESTHER : Herman_2006_J.Clin.Endocrinol.Metab_91_4612 |
| PubMedSearch : Herman_2006_J.Clin.Endocrinol.Metab_91_4612 |
| PubMedID: 16912128 |
| Title : Tolerability and pharmacokinetics of metformin and the dipeptidyl peptidase-4 inhibitor sitagliptin when co-administered in patients with type 2 diabetes - Herman_2006_Curr.Med.Res.Opin_22_1939 |
| Author(s) : Herman GA , Bergman A , Yi B , Kipnes M |
| Ref : Curr Med Res Opin , 22 :1939 , 2006 |
| Abstract : Herman_2006_Curr.Med.Res.Opin_22_1939 |
| ESTHER : Herman_2006_Curr.Med.Res.Opin_22_1939 |
| PubMedSearch : Herman_2006_Curr.Med.Res.Opin_22_1939 |
| PubMedID: 17022853 |
| Title : Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers - Bergman_2006_Clin.Ther_28_55 |
| Author(s) : Bergman AJ , Stevens C , Zhou Y , Yi B , Laethem M , De Smet M , Snyder K , Hilliard D , Tanaka W , Zeng W , Tanen M , Wang AQ , Chen L , Winchell G , Davies MJ , Ramael S , Wagner JA , Herman GA |
| Ref : Clin Ther , 28 :55 , 2006 |
| Abstract : Bergman_2006_Clin.Ther_28_55 |
| ESTHER : Bergman_2006_Clin.Ther_28_55 |
| PubMedSearch : Bergman_2006_Clin.Ther_28_55 |
| PubMedID: 16490580 |
| Title : Sitagliptin: a dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes - Miller_2006_Ann.Pharmacother_40_1336 |
| Author(s) : Miller S , St Onge EL |
| Ref : Annals of Pharmacotherapy , 40 :1336 , 2006 |
| Abstract : Miller_2006_Ann.Pharmacother_40_1336 |
| ESTHER : Miller_2006_Ann.Pharmacother_40_1336 |
| PubMedSearch : Miller_2006_Ann.Pharmacother_40_1336 |
| PubMedID: 16868220 |
| Title : Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus - Raz_2006_Diabetologia_49_2564 |
| Author(s) : Raz I , Hanefeld M , Xu L , Caria C , Williams-Herman D , Khatami H |
| Ref : Diabetologia , 49 :2564 , 2006 |
| Abstract : Raz_2006_Diabetologia_49_2564 |
| ESTHER : Raz_2006_Diabetologia_49_2564 |
| PubMedSearch : Raz_2006_Diabetologia_49_2564 |
| PubMedID: 17001471 |
| Title : Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study - Rosenstock_2006_Clin.Ther_28_1556 |
| Author(s) : Rosenstock J , Brazg R , Andryuk PJ , Lu K , Stein P |
| Ref : Clin Ther , 28 :1556 , 2006 |
| Abstract : Rosenstock_2006_Clin.Ther_28_1556 |
| ESTHER : Rosenstock_2006_Clin.Ther_28_1556 |
| PubMedSearch : Rosenstock_2006_Clin.Ther_28_1556 |
| PubMedID: 17157112 |
| Title : Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes - Mu_2006_Diabetes_55_1695 |
| Author(s) : Mu J , Woods J , Zhou YP , Roy RS , Li Z , Zycband E , Feng Y , Zhu L , Li C , Howard AD , Moller DE , Thornberry NA , Zhang BB |
| Ref : Diabetes , 55 :1695 , 2006 |
| Abstract : Mu_2006_Diabetes_55_1695 |
| ESTHER : Mu_2006_Diabetes_55_1695 |
| PubMedSearch : Mu_2006_Diabetes_55_1695 |
| PubMedID: 16731832 |
| Title : (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)- yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes - Kim_2005_J.Med.Chem_48_141 |
| Author(s) : Kim D , Wang L , Beconi M , Eiermann GJ , Fisher MH , He H , Hickey GJ , Kowalchick JE , Leiting B , Lyons K , Marsilio F , McCann ME , Patel RA , Petrov A , Scapin G , Patel SB , Roy RS , Wu JK , Wyvratt MJ , Zhang BB , Zhu L , Thornberry NA , Weber AE |
| Ref : Journal of Medicinal Chemistry , 48 :141 , 2005 |
| Abstract : Kim_2005_J.Med.Chem_48_141 |
| ESTHER : Kim_2005_J.Med.Chem_48_141 |
| PubMedSearch : Kim_2005_J.Med.Chem_48_141 |
| PubMedID: 15634008 |
| Gene_locus related to this paper: human-DPP4 |