Prochlorperazine
Prochlorperazine and (trifluoperazine) stimulated the rate of acetylthiocholine (ATCh) hydrolysis by AChE (Katz et al. 2018). Prochlorperazine is a synthetic propylpiperazine derivative of phenothiazine with antiemetic, antipsychotic, antihistaminic, and anticholinergic activities. Prochlorperazine antagonizes the dopamine D2-receptor in the chemoreceptor trigger zone (CTZ) of the brain and may prevent chemotherapy-induced emesis
General
Type : Phenothiazine,Sulfur Compound,Not A\/B H target
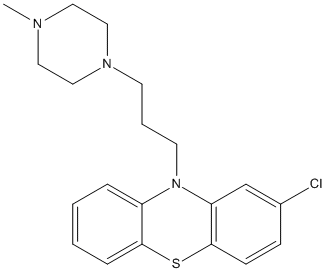
Chemical_Nomenclature : 2-chloro-10-[3-(4-methylpiperazin-1-yl)propyl]phenothiazine
Canonical SMILES : CN1CCN(CC1)CCCN2C3=CC=CC=C3SC4=C2C=C(C=C4)Cl
InChI : InChI=1S\/C20H24ClN3S\/c1-22-11-13-23(14-12-22)9-4-10-24-17-5-2-3-6-19(17)25-20-8-7-16(21)15-18(20)24\/h2-3,5-8,15H,4,9-14H2,1H3
InChIKey : WIKYUJGCLQQFNW-UHFFFAOYSA-N
Other name(s) : Prochlorperazin,Prochlorpromazine,Chlormeprazine
MW : 373.9
Formula : C20H24ClN3S
CAS_number : 58-38-8
PubChem : 4917
UniChem : WIKYUJGCLQQFNW-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families :
Stucture :
Protein :
References (6)
| Title : Antiviral activity of chlorpromazine, fluphenazine, perphenazine, prochlorperazine, and thioridazine towards RNA-viruses. A review - Otreba_2020_Eur.J.Pharmacol_887_173553 |
| Author(s) : Otreba M , Kosmider L , Rzepecka-Stojko A |
| Ref : European Journal of Pharmacology , 887 :173553 , 2020 |
| Abstract : Otreba_2020_Eur.J.Pharmacol_887_173553 |
| ESTHER : Otreba_2020_Eur.J.Pharmacol_887_173553 |
| PubMedSearch : Otreba_2020_Eur.J.Pharmacol_887_173553 |
| PubMedID: 32949606 |
| Title : Characterization of binding of antipsychotics to muscarinic receptors using mouse cerebral cortex - Obara_2019_J.Pharmacol.Sci_140_197 |
| Author(s) : Obara K , Horiguchi S , Shimada T , Ikarashi T , Yamaki F , Matsuo K , Yoshio T , Tanaka Y |
| Ref : J Pharmacol Sci , 140 :197 , 2019 |
| Abstract : Obara_2019_J.Pharmacol.Sci_140_197 |
| ESTHER : Obara_2019_J.Pharmacol.Sci_140_197 |
| PubMedSearch : Obara_2019_J.Pharmacol.Sci_140_197 |
| PubMedID: 31178327 |
| Title : New therapeutic approaches and novel alternatives for organophosphate toxicity - Katz_2018_Toxicol.Lett_291_1 |
| Author(s) : Katz FS , Pecic S , Schneider L , Zhu Z , Hastings A , Luzac M , MacDonald J , Landry DW , Stojanovic MN |
| Ref : Toxicol Lett , 291 :1 , 2018 |
| Abstract : Katz_2018_Toxicol.Lett_291_1 |
| ESTHER : Katz_2018_Toxicol.Lett_291_1 |
| PubMedSearch : Katz_2018_Toxicol.Lett_291_1 |
| PubMedID: 29614332 |
| Title : Effects of diverse psychopharmacological substances on the activity of brain prolyl oligopeptidase - Peltonen_2011_Basic.Clin.Pharmacol.Toxicol_108_46 |
| Author(s) : Peltonen I , Mannisto PT |
| Ref : Basic Clin Pharmacol Toxicol , 108 :46 , 2011 |
| Abstract : Peltonen_2011_Basic.Clin.Pharmacol.Toxicol_108_46 |
| ESTHER : Peltonen_2011_Basic.Clin.Pharmacol.Toxicol_108_46 |
| PubMedSearch : Peltonen_2011_Basic.Clin.Pharmacol.Toxicol_108_46 |
| PubMedID: 20825390 |
| Title : In vitro human plasma leucine(5)-enkephalin degradation is inhibited by a select number of drugs with the phenothiazine molecule in their chemical structure - Mosnaim_2003_Pharmacology_67_6 |
| Author(s) : Mosnaim AD , Puente J , Saavedra R , Diamond S , Wolf ME |
| Ref : Pharmacology , 67 :6 , 2003 |
| Abstract : Mosnaim_2003_Pharmacology_67_6 |
| ESTHER : Mosnaim_2003_Pharmacology_67_6 |
| PubMedSearch : Mosnaim_2003_Pharmacology_67_6 |
| PubMedID: 12444298 |
| Title : Inhibition of butyrylcholinesterase by phenothiazine derivatives - Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| Author(s) : Debord J , Merle L , Bollinger JC , Dantoine T |
| Ref : J Enzyme Inhib Med Chem , 17 :197 , 2002 |
| Abstract : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| ESTHER : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| PubMedSearch : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| PubMedID: 12443046 |