MmPPOX
IC50 around 250 nM. for LIPE but not very specific FAAH (IC50 6 nM) and MAGL (IC50 0.4 microM); hBAT5 (human-ABHD16A) 1-LG hydrolysis (IC50 8.3 nM
General
Type : Oxadiazolone,Oxadiazol,Lipase inhibitor,Phenoxyphenyl
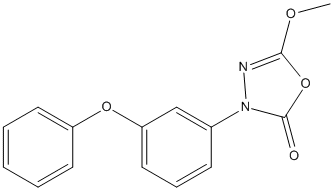
Chemical_Nomenclature : 5-methoxy-3-(3-phenoxyphenyl)-1,3,4-oxadiazol-2-one
Canonical SMILES : COC1=NN(C(=O)O1)C2=CC(=CC=C2)OC3=CC=CC=C3
InChI : InChI=1S\/C15H12N2O4\/c1-19-14-16-17(15(18)21-14)11-6-5-9-13(10-11)20-12-7-3-2-4-8-12\/h2-10H,1H3
InChIKey : MAWQNKAJQQKDTD-UHFFFAOYSA-N
Other name(s) : SCHEMBL6551376,CHEMBL2171701,5-Methoxy-3-(3-phenoxyphenyl)-1,3,4-oxadiazol-2(3H)-one,Compound-7600,5-methoxy-3-(3-phenoxyphenyl)-3H-[1,3,4]oxadiazol-2-one,Compound 7600,C7600,BDBM50450755
MW : 284.27
Formula : C15H12N2O4
CAS_number :
PubChem : 18184779
UniChem : MAWQNKAJQQKDTD-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : MmPPOX ligand of proteins in family: Acidic_Lipase || Hormone-sensitive_lipase_like || ABHD16
Stucture :
Protein : human-LIPE || human-ABHD16A
References (7)
| Title : Biochemical and Pharmacological Characterization of the Human Lymphocyte Antigen B-Associated Transcript 5 (BAT5\/ABHD16A) - Savinainen_2014_PLoS.One_9_e109869 |
| Author(s) : Savinainen JR , Patel JZ , Parkkari T , Navia-Paldanius D , Marjamaa JJ , Laitinen T , Nevalainen T , Laitinen JT |
| Ref : PLoS ONE , 9 :e109869 , 2014 |
| Abstract : Savinainen_2014_PLoS.One_9_e109869 |
| ESTHER : Savinainen_2014_PLoS.One_9_e109869 |
| PubMedSearch : Savinainen_2014_PLoS.One_9_e109869 |
| PubMedID: 25290914 |
| Gene_locus related to this paper: human-ABHD16A , mouse-Abhd16a |
| Title : The molecular mechanism of human hormone-sensitive lipase inhibition by substituted 3-phenyl-5-alkoxy-1,3,4-oxadiazol-2-ones - Ben Ali_2012_Biochimie_94_137 |
| Author(s) : Ben Ali Y , Verger R , Carriere F , Petry S , Muller G , Abousalham A |
| Ref : Biochimie , 94 :137 , 2012 |
| Abstract : Ben Ali_2012_Biochimie_94_137 |
| ESTHER : Ben Ali_2012_Biochimie_94_137 |
| PubMedSearch : Ben Ali_2012_Biochimie_94_137 |
| PubMedID: 22008857 |
| Gene_locus related to this paper: human-LIPE |
| Title : MmPPOX inhibits Mycobacterium tuberculosis lipolytic enzymes belonging to the hormone-sensitive lipase family and alters mycobacterial growth - Delorme_2012_PLoS.One_7_e46493 |
| Author(s) : Delorme V , Diomande SV , Dedieu L , Cavalier JF , Carriere F , Kremer L , Leclaire J , Fotiadu F , Canaan S |
| Ref : PLoS ONE , 7 :e46493 , 2012 |
| Abstract : Delorme_2012_PLoS.One_7_e46493 |
| ESTHER : Delorme_2012_PLoS.One_7_e46493 |
| PubMedSearch : Delorme_2012_PLoS.One_7_e46493 |
| PubMedID: 23029536 |
| Gene_locus related to this paper: myctu-Rv2970c |
| Title : Analysis of the discriminative inhibition of mammalian digestive lipases by 3-phenyl substituted 1,3,4-oxadiazol-2(3H)-ones. - Point_2012_Eur.J.Med.Chem_58_452 |
| Author(s) : Point V , Pavan Kumar KV , Marc S , Delorme V , Parsiegla G , Amara S , Carriere F , Buono G , Fotiadu F , Canaan S , Leclaire J , Cavalier JF |
| Ref : Eur Journal of Medicinal Chemistry , 58 :452 , 2012 |
| Abstract : Point_2012_Eur.J.Med.Chem_58_452 |
| ESTHER : Point_2012_Eur.J.Med.Chem_58_452 |
| PubMedSearch : Point_2012_Eur.J.Med.Chem_58_452 |
| PubMedID: 23153815 |
| Gene_locus related to this paper: canfa-1lipg , cavpo-2plrp , human-CEL , human-PNLIP , human-PNLIPRP2 , pig-1plip |
| Title : Characterization of binding properties of monoglyceride lipase inhibitors by a versatile fluorescence-based technique - Savinainen_2010_Anal.Biochem_399_132 |
| Author(s) : Savinainen JR , Yoshino M , Minkkila A , Nevalainen T , Laitinen JT |
| Ref : Analytical Biochemistry , 399 :132 , 2010 |
| Abstract : Savinainen_2010_Anal.Biochem_399_132 |
| ESTHER : Savinainen_2010_Anal.Biochem_399_132 |
| PubMedSearch : Savinainen_2010_Anal.Biochem_399_132 |
| PubMedID: 20005861 |
| Gene_locus related to this paper: human-MGLL |
| Title : Screening of various hormone-sensitive lipase inhibitors as endocannabinoid-hydrolyzing enzyme inhibitors - |
| Author(s) : Minkkila A , Savinainen JR , Kasnanen H , Xhaard H , Nevalainen T , Laitinen JT , Poso A , Leppanen J , Saario SM |
| Ref : ChemMedChem , 4 :1253 , 2009 |
| PubMedID: 19472270 |
| Title : Use of an inhibitor to identify members of the hormone-sensitive lipase family - Ben Ali_2006_Biochemistry_45_14183 |
| Author(s) : Ben Ali Y , Chahinian H , Petry S , Muller G , Lebrun R , Verger R , Carriere F , Mandrich L , Rossi M , Manco G , Sarda L , Abousalham A |
| Ref : Biochemistry , 45 :14183 , 2006 |
| Abstract : Ben Ali_2006_Biochemistry_45_14183 |
| ESTHER : Ben Ali_2006_Biochemistry_45_14183 |
| PubMedSearch : Ben Ali_2006_Biochemistry_45_14183 |
| PubMedID: 17115713 |
| Gene_locus related to this paper: human-LIPE |