Irinotecan
Irinotecan (CPT-11), an anticancer prodrug is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to its active metabolite SN-38 (7-ethyl-10-hydroxycamptothecin) a potent inhibitor of topoisomerase I. CPT-11 is hydrolysed by carboxylesterases in human pig and rabbit. Horse BChE hydrolyses 200 times faster CPT-11 than human BChE
General
Type : Carbamate,Drug,Derivative of Irinotecan,Not A\/B H target
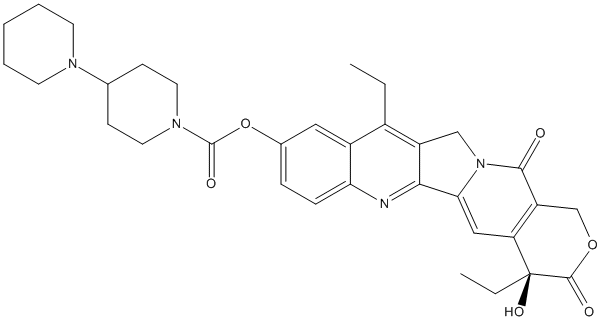
Chemical_Nomenclature : 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin) || [(19S)-10,19-diethyl-19-hydroxy-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaen-7-yl] 4-piperidin-1-ylpiperidine-1-carboxylate
Canonical SMILES : CCC1=C2C=C(C=CC2=NC3=C1CN4C3=CC5=C(C4=O)COC(=O)C5(CC)O)OC(=O)N6CCC(CC6)N7CCCCC7
InChI : InChI=1S\/C33H38N4O6\/c1-3-22-23-16-21(43-32(40)36-14-10-20(11-15-36)35-12-6-5-7-13-35)8-9-27(23)34-29-24(22)18-37-28(29)17-26-25(30(37)38)19-42-31(39)33(26,41)4-2\/h8-9,16-17,20,41H,3-7,10-15,18-19H2,1-2H3\/t33-\/m0\/s1
InChIKey : UWKQSNNFCGGAFS-XIFFEERXSA-N
Other name(s) : CPT-11,Camptosar,Irinotecanum,Biotecan
MW : 586.69
Formula : C33H38N4O6
CAS_number : 97682-44-5
PubChem : 60838
UniChem : UWKQSNNFCGGAFS-XIFFEERXSA-N
IUPHAR : 6823
Wikipedia : Irinotecan
Target
Families : Irinotecan ligand of proteins in family: Carboxylesterase || Carb_B_Chordata || Carb_B_Bacteria
Stucture : 1U65 Torpedo acetylcholinesterase complex of the anticancer prodrug CPT-11 Irinotecan
Protein : rabit-1cxes
References (24)
| Title : Crystal structure of the Geobacillus stearothermophilus carboxylesterase Est55 and its activation of prodrug CPT-11 - Liu_2007_J.Mol.Biol_367_212 |
| Author(s) : Liu P , Ewis HE , Tai PC , Lu CD , Weber IT |
| Ref : Journal of Molecular Biology , 367 :212 , 2007 |
| Abstract : Liu_2007_J.Mol.Biol_367_212 |
| ESTHER : Liu_2007_J.Mol.Biol_367_212 |
| PubMedSearch : Liu_2007_J.Mol.Biol_367_212 |
| PubMedID: 17239398 |
| Gene_locus related to this paper: geost-est50 |
| Title : The 3D structure of the anticancer prodrug CPT-11 with Torpedo californica acetylcholinesterase rationalizes its inhibitory action on AChE and its hydrolysis by butyrylcholinesterase and carboxylesterase - Harel_2005_Chem.Biol.Interact_157-158_153 |
| Author(s) : Harel M , Hyatt JL , Brumshtein B , Morton CL , Wadkins RM , Silman I , Sussman JL , Potter PM |
| Ref : Chemico-Biological Interactions , 157-158 :153 , 2005 |
| Abstract : Harel_2005_Chem.Biol.Interact_157-158_153 |
| ESTHER : Harel_2005_Chem.Biol.Interact_157-158_153 |
| PubMedSearch : Harel_2005_Chem.Biol.Interact_157-158_153 |
| PubMedID: 16289500 |
| Gene_locus related to this paper: torca-ACHE |
| Title : The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action - Harel_2005_Mol.Pharmacol_67_1874 |
| Author(s) : Harel M , Hyatt JL , Brumshtein B , Morton CL , Yoon KJ , Wadkins RM , Silman I , Sussman JL , Potter PM |
| Ref : Molecular Pharmacology , 67 :1874 , 2005 |
| Abstract : Harel_2005_Mol.Pharmacol_67_1874 |
| ESTHER : Harel_2005_Mol.Pharmacol_67_1874 |
| PubMedSearch : Harel_2005_Mol.Pharmacol_67_1874 |
| PubMedID: 15772291 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Inhibition of acetylcholinesterase by the anticancer prodrug CPT-11 - Hyatt_2005_Chem.Biol.Interact_157-158_247 |
| Author(s) : Hyatt JL , Tsurkan L , Morton CL , Yoon KJ , Harel M , Brumshtein B , Silman I , Sussman JL , Wadkins RM , Potter PM |
| Ref : Chemico-Biological Interactions , 157-158 :247 , 2005 |
| Abstract : Hyatt_2005_Chem.Biol.Interact_157-158_247 |
| ESTHER : Hyatt_2005_Chem.Biol.Interact_157-158_247 |
| PubMedSearch : Hyatt_2005_Chem.Biol.Interact_157-158_247 |
| PubMedID: 16257398 |
| Title : Lessons learned from the irinotecan metabolic pathway - Ma_2003_Curr.Med.Chem_10_41 |
| Author(s) : Ma MK , McLeod HL |
| Ref : Curr Med Chem , 10 :41 , 2003 |
| Abstract : Ma_2003_Curr.Med.Chem_10_41 |
| ESTHER : Ma_2003_Curr.Med.Chem_10_41 |
| PubMedSearch : Ma_2003_Curr.Med.Chem_10_41 |
| PubMedID: 12570720 |
| Title : Intracellular conversion of irinotecan to its active form, SN-38, by native carboxylesterase in human non-small cell lung cancer - Ohtsuka_2003_Lung.Cancer_41_187 |
| Author(s) : Ohtsuka K , Inoue S , Kameyama M , Kanetoshi A , Fujimoto T , Takaoka K , Araya Y , Shida A |
| Ref : Lung Cancer , 41 :187 , 2003 |
| Abstract : Ohtsuka_2003_Lung.Cancer_41_187 |
| ESTHER : Ohtsuka_2003_Lung.Cancer_41_187 |
| PubMedSearch : Ohtsuka_2003_Lung.Cancer_41_187 |
| PubMedID: 12871782 |
| Title : The relative contributions of carboxylesterase and beta-glucuronidase in the formation of SN-38 in human colorectal tumours - Tobin_2003_Oncol.Rep_10_1977 |
| Author(s) : Tobin PJ , Dodds HM , Clarke S , Schnitzler M , Rivory LP |
| Ref : Oncol Rep , 10 :1977 , 2003 |
| Abstract : Tobin_2003_Oncol.Rep_10_1977 |
| ESTHER : Tobin_2003_Oncol.Rep_10_1977 |
| PubMedSearch : Tobin_2003_Oncol.Rep_10_1977 |
| PubMedID: 14534729 |
| Title : A new metabolite of irinotecan in which formation is mediated by human hepatic cytochrome P-450 3A4 - Sai_2001_Drug.Metab.Dispos_29_1505 |
| Author(s) : Sai K , Kaniwa N , Ozawa S , Sawada JI |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 29 :1505 , 2001 |
| Abstract : Sai_2001_Drug.Metab.Dispos_29_1505 |
| ESTHER : Sai_2001_Drug.Metab.Dispos_29_1505 |
| PubMedSearch : Sai_2001_Drug.Metab.Dispos_29_1505 |
| PubMedID: 11602529 |
| Title : Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans - Santos_2000_Clin.Cancer.Res_6_2012 |
| Author(s) : Santos A , Zanetta S , Cresteil T , Deroussent A , Pein F , Raymond E , Vernillet L , Risse ML , Boige V , Gouyette A , Vassal G |
| Ref : Clin Cancer Research , 6 :2012 , 2000 |
| Abstract : Santos_2000_Clin.Cancer.Res_6_2012 |
| ESTHER : Santos_2000_Clin.Cancer.Res_6_2012 |
| PubMedSearch : Santos_2000_Clin.Cancer.Res_6_2012 |
| PubMedID: 10815927 |
| Title : Isolation and characterization of a cDNA encoding a horse liver butyrylcholinesterase: evidence for CPT-11 drug activation - Wierdl_2000_Biochem.Pharmacol_59_773 |
| Author(s) : Wierdl M , Morton CL , Danks MK , Potter PM |
| Ref : Biochemical Pharmacology , 59 :773 , 2000 |
| Abstract : Wierdl_2000_Biochem.Pharmacol_59_773 |
| ESTHER : Wierdl_2000_Biochem.Pharmacol_59_773 |
| PubMedSearch : Wierdl_2000_Biochem.Pharmacol_59_773 |
| PubMedID: 10718335 |
| Gene_locus related to this paper: horse-BCHE |
| Title : The anticancer prodrug CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase - Morton_1999_Cancer.Res_59_1458 |
| Author(s) : Morton CL , Wadkins RM , Danks MK , Potter PM |
| Ref : Cancer Research , 59 :1458 , 1999 |
| Abstract : Morton_1999_Cancer.Res_59_1458 |
| ESTHER : Morton_1999_Cancer.Res_59_1458 |
| PubMedSearch : Morton_1999_Cancer.Res_59_1458 |
| PubMedID: 10197614 |
| Title : The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11) - Dodds_1999_Mol.Pharmacol_56_1346 |
| Author(s) : Dodds HM , Rivory LP |
| Ref : Molecular Pharmacology , 56 :1346 , 1999 |
| Abstract : Dodds_1999_Mol.Pharmacol_56_1346 |
| ESTHER : Dodds_1999_Mol.Pharmacol_56_1346 |
| PubMedSearch : Dodds_1999_Mol.Pharmacol_56_1346 |
| PubMedID: 10570064 |
| Title : Water soluble 20(S)-glycinate esters of 10,11-methylenedioxycamptothecins are highly active against human breast cancer xenografts - Wadkins_1999_Cancer.Res_59_3424 |
| Author(s) : Wadkins RM , Potter PM , Vladu B , Marty J , Mangold G , Weitman S , Manikumar G , Wani MC , Wall ME , Von Hoff DD |
| Ref : Cancer Research , 59 :3424 , 1999 |
| Abstract : Wadkins_1999_Cancer.Res_59_3424 |
| ESTHER : Wadkins_1999_Cancer.Res_59_3424 |
| PubMedSearch : Wadkins_1999_Cancer.Res_59_3424 |
| PubMedID: 10416605 |
| Title : Conversion of the CPT-11 metabolite APC to SN-38 by rabbit liver carboxylesterase - Guichard_1998_Clin.Cancer.Res_4_3089 |
| Author(s) : Guichard SM , Morton CL , Krull EJ , Stewart CF , Danks MK , Potter PM |
| Ref : Clin Cancer Research , 4 :3089 , 1998 |
| Abstract : Guichard_1998_Clin.Cancer.Res_4_3089 |
| ESTHER : Guichard_1998_Clin.Cancer.Res_4_3089 |
| PubMedSearch : Guichard_1998_Clin.Cancer.Res_4_3089 |
| PubMedID: 9865925 |
| Title : Gastrointestinal toxicity or irinotecan - Hecht_1998_Oncology_12_72 |
| Author(s) : Hecht JR |
| Ref : Oncology , 12 :72 , 1998 |
| Abstract : Hecht_1998_Oncology_12_72 |
| ESTHER : Hecht_1998_Oncology_12_72 |
| PubMedSearch : Hecht_1998_Oncology_12_72 |
| PubMedID: 9726096 |
| Title : Pharmacology of irinotecan - Kuhn_1998_Oncology_12_39 |
| Author(s) : Kuhn JG |
| Ref : Oncology , 12 :39 , 1998 |
| Abstract : Kuhn_1998_Oncology_12_39 |
| ESTHER : Kuhn_1998_Oncology_12_39 |
| PubMedSearch : Kuhn_1998_Oncology_12_39 |
| PubMedID: 9726089 |
| Title : Cellular localization domains of a rabbit and a human carboxylesterase: influence on irinotecan (CPT-11) metabolism by the rabbit enzyme - Potter_1998_Cancer.Res_58_3627 |
| Author(s) : Potter PM , Wolverton JS , Morton CL , Wierdl M , Danks MK |
| Ref : Cancer Research , 58 :3627 , 1998 |
| Abstract : Potter_1998_Cancer.Res_58_3627 |
| ESTHER : Potter_1998_Cancer.Res_58_3627 |
| PubMedSearch : Potter_1998_Cancer.Res_58_3627 |
| PubMedID: 9721871 |
| Title : Overexpression of a rabbit liver carboxylesterase sensitizes human tumor cells to CPT-11 - Danks_1998_Cancer.Res_58_20 |
| Author(s) : Danks MK , Morton CL , Pawlik CA , Potter PM |
| Ref : Cancer Research , 58 :20 , 1998 |
| Abstract : Danks_1998_Cancer.Res_58_20 |
| ESTHER : Danks_1998_Cancer.Res_58_20 |
| PubMedSearch : Danks_1998_Cancer.Res_58_20 |
| PubMedID: 9426050 |
| Gene_locus related to this paper: rabit-1cxes |
| Title : Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug irinotecan (CPT-11) - Potter_1998_Cancer.Res_58_2646 |
| Author(s) : Potter PM , Pawlik CA , Morton CL , Naeve CW , Danks MK |
| Ref : Cancer Research , 58 :2646 , 1998 |
| Abstract : Potter_1998_Cancer.Res_58_2646 |
| ESTHER : Potter_1998_Cancer.Res_58_2646 |
| PubMedSearch : Potter_1998_Cancer.Res_58_2646 |
| PubMedID: 9635592 |
| Gene_locus related to this paper: rabit-1cxes |
| Title : The transformation of irinotecan (CPT-11) to its active metabolite SN-38 by human liver microsomes. Differential hydrolysis for the lactone and carboxylate forms - Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| Author(s) : Haaz MC , Rivory LP , Riche C , Robert J |
| Ref : Naunyn Schmiedebergs Arch Pharmacol , 356 :257 , 1997 |
| Abstract : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| ESTHER : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| PubMedSearch : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| PubMedID: 9272733 |
| Title : Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions - Slatter_1997_Drug.Metab.Dispos_25_1157 |
| Author(s) : Slatter JG , Su P , Sams JP , Schaaf LJ , Wienkers LC |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 25 :1157 , 1997 |
| Abstract : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| ESTHER : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| PubMedSearch : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| PubMedID: 9321519 |
| Title : CPT-11 in human colon-cancer cell lines and xenografts: characterization of cellular sensitivity determinants - Jansen_1997_Int.J.Cancer_70_335 |
| Author(s) : Jansen WJ , Zwart B , Hulscher ST , Giaccone G , Pinedo HM , Boven E |
| Ref : International Journal of Cancer , 70 :335 , 1997 |
| Abstract : Jansen_1997_Int.J.Cancer_70_335 |
| ESTHER : Jansen_1997_Int.J.Cancer_70_335 |
| PubMedSearch : Jansen_1997_Int.J.Cancer_70_335 |
| PubMedID: 9033637 |
| Title : Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase - Rivory_1996_Biochem.Pharmacol_52_1103 |
| Author(s) : Rivory LP , Bowles MR , Robert J , Pond SM |
| Ref : Biochemical Pharmacology , 52 :1103 , 1996 |
| Abstract : Rivory_1996_Biochem.Pharmacol_52_1103 |
| ESTHER : Rivory_1996_Biochem.Pharmacol_52_1103 |
| PubMedSearch : Rivory_1996_Biochem.Pharmacol_52_1103 |
| PubMedID: 8831730 |
| Title : Identification and properties of a major plasma metabolite of irinotecan (CPT-11) isolated from the plasma of patients - Rivory_1996_Cancer.Res_56_3689 |
| Author(s) : Rivory LP , Riou JF , Haaz MC , Sable S , Vuilhorgne M , Commercon A , Pond SM , Robert J |
| Ref : Cancer Research , 56 :3689 , 1996 |
| Abstract : Rivory_1996_Cancer.Res_56_3689 |
| ESTHER : Rivory_1996_Cancer.Res_56_3689 |
| PubMedSearch : Rivory_1996_Cancer.Res_56_3689 |
| PubMedID: 8706009 |