HuprineX
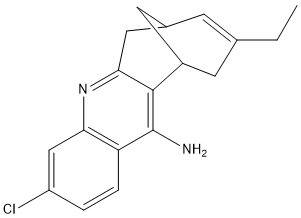
Carbobicyclic substructure of huperzine A, with the 4-aminoquinoline substructure of tacrine
General
Type : Derivative of Huperzine,Derivative of Tacrine,Quinoline,Natural_modified
Chemical_Nomenclature : (-)-12-amino-3-chloro-9-ethyl-6,7,10,11-tetrahydro-7,11-methanocycloocta[b]quinoline hydrochloride
Canonical SMILES : CCC1=CC2CC(C1)C3=C(C4=C(C=C(C=C4)Cl)N=C3C2)N
InChI : InChI=1S\/C18H19ClN2\/c1-2-10-5-11-7-12(6-10)17-16(8-11)21-15-9-13(19)3-4-14(15)18(17)20\/h3-5,9,11-12H,2,6-8H2,1H3,(H2,20,21)
InChIKey : QTPHSDHUHXUYFE-UHFFFAOYSA-N
Other name(s) : Rac-huprine H7,Huprine x,CHEMBL143812,3-chloro-9-ethyl-6,7,10,11-tetrahydro-7,11-methanocycloocta[b]quinolin-12-amine,AC1L1GD8,SCHEMBL7115053
MW : 298.81
Formula : C18H19N2Cl
CAS_number :
PubChem : 3632
UniChem : QTPHSDHUHXUYFE-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : HuprineX ligand of proteins in family: ACHE
Stucture : 1E66 Acetylcholinesterase complexed with (-)-Huprine X at 2.1A resolution
Protein : torca-ACHE
References (9)
| Title : Huprine X Attenuates The Neurotoxicity Induced by Kainic Acid, Especially Brain Inflammation - Relat_2018_Basic.Clin.Pharmacol.Toxicol_122_94 |
| Author(s) : Relat J , Perez B , Camps P , Munoz-Torrero D , Badia A , Victoria Clos M |
| Ref : Basic Clin Pharmacol Toxicol , 122 :94 , 2018 |
| Abstract : Relat_2018_Basic.Clin.Pharmacol.Toxicol_122_94 |
| ESTHER : Relat_2018_Basic.Clin.Pharmacol.Toxicol_122_94 |
| PubMedSearch : Relat_2018_Basic.Clin.Pharmacol.Toxicol_122_94 |
| PubMedID: 28724203 |
| Title : Huprine X and Huperzine A Improve Cognition and Regulate Some Neurochemical Processes Related with Alzheimer's Disease in Triple Transgenic Mice (3xTg-AD) - Ratia_2013_Neurodegener.Dis_11_129 |
| Author(s) : Ratia M , Gimenez-Llort L , Camps P , Munoz-Torrero D , Perez B , Clos MV , Badia A |
| Ref : Neurodegener Dis , 11 :129 , 2013 |
| Abstract : Ratia_2013_Neurodegener.Dis_11_129 |
| ESTHER : Ratia_2013_Neurodegener.Dis_11_129 |
| PubMedSearch : Ratia_2013_Neurodegener.Dis_11_129 |
| PubMedID: 22626981 |
| Title : Effect of huprine X on beta-amyloid, synaptophysin and alpha7 neuronal nicotinic acetylcholine receptors in the brain of 3xTg-AD and APPswe transgenic mice - Hedberg_2010_Neurodegener.Dis_7_379 |
| Author(s) : Hedberg MM , Clos MV , Ratia M , Gonzalez D , Lithner CU , Camps P , Munoz-Torrero D , Badia A , Gimenez-Llort L , Nordberg A |
| Ref : Neurodegener Dis , 7 :379 , 2010 |
| Abstract : Hedberg_2010_Neurodegener.Dis_7_379 |
| ESTHER : Hedberg_2010_Neurodegener.Dis_7_379 |
| PubMedSearch : Hedberg_2010_Neurodegener.Dis_7_379 |
| PubMedID: 20689242 |
| Title : Nicotinic-receptor potentiator drugs, huprine X and galantamine, increase ACh release by blocking AChE activity but not acting on nicotinic receptors - Roman_2005_Brain.Res_1061_73 |
| Author(s) : Roman S , Badia A , Camps P , Munoz-Torrero D , Clos MV |
| Ref : Brain Research , 1061 :73 , 2005 |
| Abstract : Roman_2005_Brain.Res_1061_73 |
| ESTHER : Roman_2005_Brain.Res_1061_73 |
| PubMedSearch : Roman_2005_Brain.Res_1061_73 |
| PubMedID: 16248990 |
| Title : Potentiation effects of (+\/-)huprine X, a new acetylcholinesterase inhibitor, on nicotinic receptors in rat cortical synaptosomes - Roman_2004_Neuropharmacol_46_95 |
| Author(s) : Roman S , Badia A , Camps P , Clos MV |
| Ref : Neuropharmacology , 46 :95 , 2004 |
| Abstract : Roman_2004_Neuropharmacol_46_95 |
| ESTHER : Roman_2004_Neuropharmacol_46_95 |
| PubMedSearch : Roman_2004_Neuropharmacol_46_95 |
| PubMedID: 14654101 |
| Title : Interaction of a new potent anticholinesterasic compound (+\/-)huprine X with muscarinic receptors in rat brain - Roman_2002_Neurosci.Lett_325_103 |
| Author(s) : Roman S , Vivas NM , Badia A , Clos MV |
| Ref : Neuroscience Letters , 325 :103 , 2002 |
| Abstract : Roman_2002_Neurosci.Lett_325_103 |
| ESTHER : Roman_2002_Neurosci.Lett_325_103 |
| PubMedSearch : Roman_2002_Neurosci.Lett_325_103 |
| PubMedID: 12044632 |
| Title : 3D structure of Torpedo californica acetylcholinesterase complexed with huprine X at 2.1 A resolution: kinetic and molecular dynamic correlates - Dvir_2002_Biochemistry_41_2970 |
| Author(s) : Dvir H , Wong DM , Harel M , Barril X , Orozco M , Luque FJ , Munoz-Torrero D , Camps P , Rosenberry TL , Silman I , Sussman JL |
| Ref : Biochemistry , 41 :2970 , 2002 |
| Abstract : Dvir_2002_Biochemistry_41_2970 |
| ESTHER : Dvir_2002_Biochemistry_41_2970 |
| PubMedSearch : Dvir_2002_Biochemistry_41_2970 |
| PubMedID: 11863435 |
| Gene_locus related to this paper: torca-ACHE |
| Title : The pharmacology of novel acetylcholinesterase inhibitors, (+\/-)- huprines Y and X, on the Torpedo electric organ - Ros_2001_Eur.J.Pharmacol_421_77 |
| Author(s) : Ros E , Aleu J , Gomez de Aranda I , Munoz-Torrero D , Camps P , Badia A , Marsal J , Solsona C |
| Ref : European Journal of Pharmacology , 421 :77 , 2001 |
| Abstract : Ros_2001_Eur.J.Pharmacol_421_77 |
| ESTHER : Ros_2001_Eur.J.Pharmacol_421_77 |
| PubMedSearch : Ros_2001_Eur.J.Pharmacol_421_77 |
| PubMedID: 11399262 |
| Title : Huprine X is a Novel High-Affinity Inhibitor of Acetylcholinesterase That Is of Interest for Treatment of Alzheimer's Disease - Camps_2000_Mol.Pharmacol_57_409 |
| Author(s) : Camps P , Cusack B , Mallender WD , Achab RE , Morral J , Munoz-Torrero D , Rosenberry TL |
| Ref : Molecular Pharmacology , 57 :409 , 2000 |
| Abstract : Camps_2000_Mol.Pharmacol_57_409 |
| ESTHER : Camps_2000_Mol.Pharmacol_57_409 |
| PubMedSearch : Camps_2000_Mol.Pharmacol_57_409 |
| PubMedID: 10648652 |