Echothiophate
A potent, long-acting irreversible cholinesterase inhibitor used as an ocular hypertensive in the treatment of glaucoma. Occasionally used for accomodative esotropia.
General
Type : Drug,Organophosphate,Sulfur Compound,Organothiophosphate,Trimethylammonium
Chemical_Nomenclature : 2-diethoxyphosphorylsulfanylethyl(trimethyl)azanium
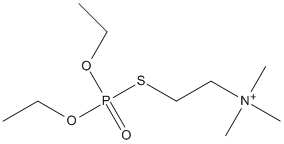
Canonical SMILES : CCOP(=O)(OCC)SCC[N+](C)(C)C
InChI : InChI=1S\/C9H23NO3PS\/c1-6-12-14(11,13-7-2)15-9-8-10(3,4)5\/h6-9H2,1-5H3\/q+1
InChIKey : BJOLKYGKSZKIGU-UHFFFAOYSA-N
Other name(s) : O,O'-diethyl S-ethyltrimethylamine phosphorothiolate iodide,(2-mercaptoethyl) trimethylammonium iodide O,O'diethylphosphorothioate,6736-03-4,Echodide,Phospholine iodide,217-MI, ecothiopate iodide,Ecothiopate,Ecothiopatum,Phospholine,UNII-0F350BVT6S,CHEBI:4753,SCHEMBL1048167,CHEMBL1201341,DB01057
MW : 383.23
Formula : C9H23INO3PS
CAS_number : 513-10-0
PubChem : 10548
UniChem : BJOLKYGKSZKIGU-UHFFFAOYSA-N
IUPHAR : 9074
Wikipedia : Echothiophate
Target
References (17)
| Title : Steady-State Kinetics of Enzyme-Catalyzed Hydrolysis of Echothiophate, a P-S Bonded Organophosphorus as Monitored by Spectrofluorimetry - Zueva_2020_Molecules_25_ |
| Author(s) : Zueva IV , Lushchekina SV , Daude D , Chabriere E , Masson P |
| Ref : Molecules , 25 : , 2020 |
| Abstract : Zueva_2020_Molecules_25_ |
| ESTHER : Zueva_2020_Molecules_25_ |
| PubMedSearch : Zueva_2020_Molecules_25_ |
| PubMedID: 32192230 |
| Title : X-ray crystallographic snapshots of reaction intermediates in the G117H mutant of human butyrylcholinesterase, a nerve agent target engineered into a catalytic bioscavenger - Nachon_2011_Biochem.J_434_73 |
| Author(s) : Nachon F , Carletti E , Wandhammer M , Nicolet Y , Schopfer LM , Masson P , Lockridge O |
| Ref : Biochemical Journal , 434 :73 , 2011 |
| Abstract : Nachon_2011_Biochem.J_434_73 |
| ESTHER : Nachon_2011_Biochem.J_434_73 |
| PubMedSearch : Nachon_2011_Biochem.J_434_73 |
| PubMedID: 21091433 |
| Gene_locus related to this paper: human-BCHE |
| Title : Role of water in aging of human butyrylcholinesterase inhibited by echothiophate: the crystal structure suggests two alternative mechanisms of aging - Nachon_2005_Biochemistry_44_1154 |
| Author(s) : Nachon F , Asojo OA , Borgstahl GE , Masson P , Lockridge O |
| Ref : Biochemistry , 44 :1154 , 2005 |
| Abstract : Nachon_2005_Biochemistry_44_1154 |
| ESTHER : Nachon_2005_Biochemistry_44_1154 |
| PubMedSearch : Nachon_2005_Biochemistry_44_1154 |
| PubMedID: 15667209 |
| Gene_locus related to this paper: human-BCHE |
| Title : Effects of organophosphates on cholinesterase activity and neurite regeneration in Aplysia - Srivatsan_1999_Chem.Biol.Interact_119-120_371 |
| Author(s) : Srivatsan M |
| Ref : Chemico-Biological Interactions , 119-120 :371 , 1999 |
| Abstract : Srivatsan_1999_Chem.Biol.Interact_119-120_371 |
| ESTHER : Srivatsan_1999_Chem.Biol.Interact_119-120_371 |
| PubMedSearch : Srivatsan_1999_Chem.Biol.Interact_119-120_371 |
| PubMedID: 10421473 |
| Title : A comparison of the electrophysiological effects of two organophosphates, mipafox and ecothiopate, on mouse limb muscles - de Blaquiere_1998_Toxicol.Appl.Pharmacol_150_350 |
| Author(s) : de Blaquiere GE , Williams FM , Blain PG , Kelly SS |
| Ref : Toxicol Appl Pharmacol , 150 :350 , 1998 |
| Abstract : de Blaquiere_1998_Toxicol.Appl.Pharmacol_150_350 |
| ESTHER : de Blaquiere_1998_Toxicol.Appl.Pharmacol_150_350 |
| PubMedSearch : de Blaquiere_1998_Toxicol.Appl.Pharmacol_150_350 |
| PubMedID: 9653066 |
| Title : Importance of aspartate-70 in organophosphate inhibition, oxime re-activation and aging of human butyrylcholinesterase - Masson_1997_Biochem.J_325_53 |
| Author(s) : Masson P , Froment MT , Bartels CF , Lockridge O |
| Ref : Biochemical Journal , 325 :53 , 1997 |
| Abstract : Masson_1997_Biochem.J_325_53 |
| ESTHER : Masson_1997_Biochem.J_325_53 |
| PubMedSearch : Masson_1997_Biochem.J_325_53 |
| PubMedID: 9224629 |
| Title : Acetylcholinesterase promotes regeneration of neurites in cultured adult neurons of Aplysia - Srivatsan_1997_Neurosci_77_921 |
| Author(s) : Srivatsan M , Peretz B |
| Ref : Neuroscience , 77 :921 , 1997 |
| Abstract : Srivatsan_1997_Neurosci_77_921 |
| ESTHER : Srivatsan_1997_Neurosci_77_921 |
| PubMedSearch : Srivatsan_1997_Neurosci_77_921 |
| PubMedID: 9070763 |
| Title : Cholinergic toxicity resulting from ocular instillation of echothiophate iodide eye drops - Manoguerra_1995_J.Toxicol.Clin.Toxicol_33_463 |
| Author(s) : Manoguerra A , Whitney C , Clark RF , Anderson B , Turchen S |
| Ref : Journal of Toxicology Clinical Toxicology , 33 :463 , 1995 |
| Abstract : Manoguerra_1995_J.Toxicol.Clin.Toxicol_33_463 |
| ESTHER : Manoguerra_1995_J.Toxicol.Clin.Toxicol_33_463 |
| PubMedSearch : Manoguerra_1995_J.Toxicol.Clin.Toxicol_33_463 |
| PubMedID: 7650771 |
| Title : Site-directed mutagenesis of active site residues reveals plasticity of human butyrylcholinesterase in substrate and inhibitor interactions - Gnatt_1994_J.Neurochem_62_749 |
| Author(s) : Gnatt A , Loewenstein Y , Yaron A , Schwarz M , Soreq H |
| Ref : Journal of Neurochemistry , 62 :749 , 1994 |
| Abstract : Gnatt_1994_J.Neurochem_62_749 |
| ESTHER : Gnatt_1994_J.Neurochem_62_749 |
| PubMedSearch : Gnatt_1994_J.Neurochem_62_749 |
| PubMedID: 8294937 |
| Title : Correlation of the anticholinesterase activity of a series of organophosphates with their ability to compete with agonist binding to muscarinic receptors - Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| Author(s) : Ward TR , Ferris DJ , Tilson HA , Mundy WR |
| Ref : Toxicology & Applied Pharmacology , 122 :300 , 1993 |
| Abstract : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| ESTHER : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| PubMedSearch : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| PubMedID: 8212012 |
| Title : Preferential inhibition of acetylcholinesterase molecular forms in rat brain - Ogane_1992_Neurochem.Res_17_489 |
| Author(s) : Ogane N , Giacobini E , Messamore E |
| Ref : Neurochemical Research , 17 :489 , 1992 |
| Abstract : Ogane_1992_Neurochem.Res_17_489 |
| ESTHER : Ogane_1992_Neurochem.Res_17_489 |
| PubMedSearch : Ogane_1992_Neurochem.Res_17_489 |
| PubMedID: 1528356 |
| Title : Electroconvulsive therapy and the chronic use of pseudocholinesterase- inhibitor (echothiophate iodide) eye drops for glaucoma. A case report - Messer_1992_Gen.Hosp.Psychiatry_14_56 |
| Author(s) : Messer GJ , Stoudemire A , Knos G , Johnson GC |
| Ref : General Hospital Psychiatry , 14 :56 , 1992 |
| Abstract : Messer_1992_Gen.Hosp.Psychiatry_14_56 |
| ESTHER : Messer_1992_Gen.Hosp.Psychiatry_14_56 |
| PubMedSearch : Messer_1992_Gen.Hosp.Psychiatry_14_56 |
| PubMedID: 1730402 |
| Title : Organophosphate increases the sensitivity of human exocrine pancreas to acetylcholine - Kandalaft_1991_Pancreas_6_398 |
| Author(s) : Kandalaft K , Liu S , Manivel C , Borner JW , Dressel TD , Sutherland DE , Goodale RL |
| Ref : Pancreas , 6 :398 , 1991 |
| Abstract : Kandalaft_1991_Pancreas_6_398 |
| ESTHER : Kandalaft_1991_Pancreas_6_398 |
| PubMedSearch : Kandalaft_1991_Pancreas_6_398 |
| PubMedID: 1715083 |
| Title : Putative M2 muscarinic receptors of rat heart have high affinity for organophosphorus anticholinesterases - Silveira_1990_Toxicol.Appl.Pharmacol_103_474 |
| Author(s) : Silveira CL , Eldefrawi AT , Eldefrawi ME |
| Ref : Toxicol Appl Pharmacol , 103 :474 , 1990 |
| Abstract : Silveira_1990_Toxicol.Appl.Pharmacol_103_474 |
| ESTHER : Silveira_1990_Toxicol.Appl.Pharmacol_103_474 |
| PubMedSearch : Silveira_1990_Toxicol.Appl.Pharmacol_103_474 |
| PubMedID: 2339420 |
| Title : Clonidine protection from soman and echothiophate toxicity in mice - Aronstam_1986_Life.Sci_39_2097 |
| Author(s) : Aronstam RS , Smith MD , Buccafusco JJ |
| Ref : Life Sciences , 39 :2097 , 1986 |
| Abstract : Aronstam_1986_Life.Sci_39_2097 |
| ESTHER : Aronstam_1986_Life.Sci_39_2097 |
| PubMedSearch : Aronstam_1986_Life.Sci_39_2097 |
| PubMedID: 3784771 |
| Title : Activation and blockade of cardiac muscarinic receptors by endogenous acetylcholine and cholinesterase inhibitors - Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| Author(s) : Brown JH , Wetzel GT , Dunlap J |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 223 :20 , 1982 |
| Abstract : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| ESTHER : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedSearch : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedID: 6288918 |
| Title : Echothiophate iodide treatment of glaucoma in pregnancy - |
| Author(s) : Birks DA , Prior VJ , Silk E , Whittaker M |
| Ref : Archives of Ophthalmology , 79 :283 , 1968 |
| PubMedID: 5640850 |