Darapladib
Darapladib is a substituted pyrimidone with inhibitory activity towards lipoprotein-associated phospholipase-A2 (Lp-PLA2), an important regulator of lipid metabolism and inflammation that circulates with lipoprotein particles and is carried into the arterial wall with low-density lipoprotein particles during the progression of atherosclerosis. Failed to demonstrate efficacy for the primary endpoints in two large phase III cardiovascular outcomes trials
General
Type : Trifluoro,Pyrimidine,Sulfur Compound
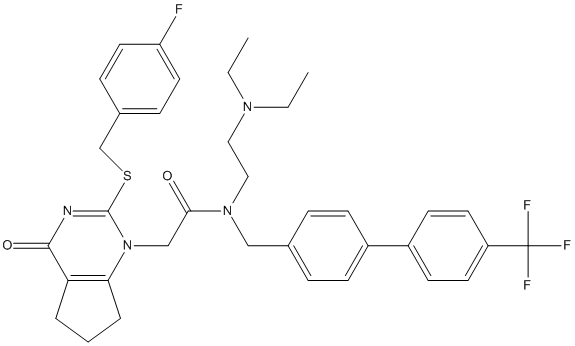
Chemical_Nomenclature : N-[2-(diethylamino)ethyl]-2-[2-[(4-fluorophenyl)methylsulfanyl]-4-oxo-6,7-dihydro-5H-cyclopenta[d]pyrimidin-1-yl]-N-[[4-[4-(trifluoromethyl)phenyl]phenyl]methyl]acetamide
Canonical SMILES : CCN(CC)CCN(CC1=CC=C(C=C1)C2=CC=C(C=C2)C(F)(F)F)C(=O)CN3C4=C(CCC4)C(=O)N=C3SCC5=CC=C(C=C5)F
InChI : InChI=1S\/C36H38F4N4O2S\/c1-3-42(4-2)20-21-43(22-25-8-12-27(13-9-25)28-14-16-29(17-15-28)36(38,39)40)33(45)23-44-32-7-5-6-31(32)34(46)41-35(44)47-24-26-10-18-30(37)19-11-26\/h8-19H,3-7,20-24H2,1-2H3
InChIKey : WDPFJWLDPVQCAJ-UHFFFAOYSA-N
Other name(s) : UNII-UI1U1MYH09,CHEMBL204021,SB-480848,SB 480848
MW : 666.77
Formula : C36H38F4N4O2S
CAS_number : 356057-34-6
PubChem : 9939609
UniChem : WDPFJWLDPVQCAJ-UHFFFAOYSA-N
IUPHAR : 6696
Wikipedia : Darapladib
Target
Families : Darapladib ligand of proteins in family: PAF-Acetylhydrolase
Stucture : 5I9I Crystal structure of human LP_PLA2 in complex with Darapladib
Protein : human-PLA2G7
References (15)
| Title : Inhibiting PLA2G7 reverses the immunosuppressive function of intratumoral macrophages and augments immunotherapy response in hepatocellular carcinoma - Zhang_2024_J.Immunother.Cancer_12_e008094 |
| Author(s) : Zhang F , Liu W , Meng F , Jiang Q , Tang W , Liu Z , Lin X , Xue R , Zhang S , Dong L |
| Ref : J Immunother Cancer , 12 : , 2024 |
| Abstract : Zhang_2024_J.Immunother.Cancer_12_e008094 |
| ESTHER : Zhang_2024_J.Immunother.Cancer_12_e008094 |
| PubMedSearch : Zhang_2024_J.Immunother.Cancer_12_e008094 |
| PubMedID: 38272562 |
| Gene_locus related to this paper: human-PLA2G7 |
| Title : The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism - Oh_2023_Nat.Commun_14_5728 |
| Author(s) : Oh M , Jang SY , Lee JY , Kim JW , Jung Y , Kim J , Seo J , Han TS , Jang E , Son HY , Kim D , Kim MW , Park JS , Song KH , Oh KJ , Kim WK , Bae KH , Huh YM , Kim SH , Han BS , Lee SC , Hwang GS , Lee EW |
| Ref : Nat Commun , 14 :5728 , 2023 |
| Abstract : Oh_2023_Nat.Commun_14_5728 |
| ESTHER : Oh_2023_Nat.Commun_14_5728 |
| PubMedSearch : Oh_2023_Nat.Commun_14_5728 |
| PubMedID: 37714840 |
| Title : Regioselectivity of thiouracil alkylation: Application to optimization of Darapladib synthesis - Guibbal_2018_Bioorg.Med.Chem.Lett_28_787 |
| Author(s) : Guibbal F , Benard S , Patche J , Meneyrol V , Couprie J , Yong-Sang J , Meilhac O , Jestin E |
| Ref : Bioorganic & Medicinal Chemistry Lett , 28 :787 , 2018 |
| Abstract : Guibbal_2018_Bioorg.Med.Chem.Lett_28_787 |
| ESTHER : Guibbal_2018_Bioorg.Med.Chem.Lett_28_787 |
| PubMedSearch : Guibbal_2018_Bioorg.Med.Chem.Lett_28_787 |
| PubMedID: 29336874 |
| Title : Pharmacogenetic meta-analysis of baseline risk factors, pharmacodynamic, efficacy and tolerability endpoints from two large global cardiovascular outcomes trials for darapladib - Yeo_2017_PLoS.One_12_e0182115 |
| Author(s) : Yeo A , Li L , Warren L , Aponte J , Fraser D , King K , Johansson K , Barnes A , Macphee C , Davies R , Chissoe S , Tarka E , O'Donoghue ML , White HD , Wallentin L , Waterworth D |
| Ref : PLoS ONE , 12 :e0182115 , 2017 |
| Abstract : Yeo_2017_PLoS.One_12_e0182115 |
| ESTHER : Yeo_2017_PLoS.One_12_e0182115 |
| PubMedSearch : Yeo_2017_PLoS.One_12_e0182115 |
| PubMedID: 28753643 |
| Gene_locus related to this paper: human-PLA2G7 |
| Title : Darapladib, a lipoprotein-associated phospholipase A2 inhibitor, in diabetic macular edema: a 3-month placebo-controlled study - Staurenghi_2015_Ophthalmology_122_990 |
| Author(s) : Staurenghi G , Ye L , Magee MH , Danis RP , Wurzelmann J , Adamson P , McLaughlin MM |
| Ref : Ophthalmology , 122 :990 , 2015 |
| Abstract : Staurenghi_2015_Ophthalmology_122_990 |
| ESTHER : Staurenghi_2015_Ophthalmology_122_990 |
| PubMedSearch : Staurenghi_2015_Ophthalmology_122_990 |
| PubMedID: 25749297 |
| Title : Atherosclerotic plaque inflammation varies between vascular sites and correlates with response to inhibition of lipoprotein-associated phospholipase A2 - Fenning_2015_J.Am.Heart.Assoc_4_ |
| Author(s) : Fenning RS , Burgert ME , Hamamdzic D , Peyster EG , Mohler ER , Kangovi S , Jucker BM , Lenhard SC , Macphee CH , Wilensky RL |
| Ref : J Am Heart Assoc , 4 : , 2015 |
| Abstract : Fenning_2015_J.Am.Heart.Assoc_4_ |
| ESTHER : Fenning_2015_J.Am.Heart.Assoc_4_ |
| PubMedSearch : Fenning_2015_J.Am.Heart.Assoc_4_ |
| PubMedID: 25672369 |
| Title : Unraveling the PAF-AH\/Lp-PLA2 controversy - Stafforini_2014_J.Lipid.Res_55_1811 |
| Author(s) : Stafforini DM , Zimmerman GA |
| Ref : J Lipid Res , 55 :1811 , 2014 |
| Abstract : Stafforini_2014_J.Lipid.Res_55_1811 |
| ESTHER : Stafforini_2014_J.Lipid.Res_55_1811 |
| PubMedSearch : Stafforini_2014_J.Lipid.Res_55_1811 |
| PubMedID: 25007789 |
| Title : To hydrolyze or not to hydrolyze: the dilemma of platelet-activating factor acetylhydrolase - Marathe_2014_J.Lipid.Res_55_1847 |
| Author(s) : Marathe GK , Pandit C , Lakshmikanth CL , Chaithra VH , Jacob SP , D'Souza CJ |
| Ref : J Lipid Res , 55 :1847 , 2014 |
| Abstract : Marathe_2014_J.Lipid.Res_55_1847 |
| ESTHER : Marathe_2014_J.Lipid.Res_55_1847 |
| PubMedSearch : Marathe_2014_J.Lipid.Res_55_1847 |
| PubMedID: 24859738 |
| Title : Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A(2) - Nagano_2013_Bioorg.Med.Chem.Lett_23_839 |
| Author(s) : Nagano JM , Hsu KL , Whitby LR , Niphakis MJ , Speers AE , Brown SJ , Spicer T , Fernandez-Vega V , Ferguson J , Hodder P , Srinivasan P , Gonzalez TD , Rosen H , Bahnson BJ , Cravatt BF |
| Ref : Bioorganic & Medicinal Chemistry Lett , 23 :839 , 2013 |
| Abstract : Nagano_2013_Bioorg.Med.Chem.Lett_23_839 |
| ESTHER : Nagano_2013_Bioorg.Med.Chem.Lett_23_839 |
| PubMedSearch : Nagano_2013_Bioorg.Med.Chem.Lett_23_839 |
| PubMedID: 23260346 |
| Title : Darapladib - Bui_2010_Expert.Opin.Investig.Drugs_19_161 |
| Author(s) : Bui QT , Wilensky RL |
| Ref : Expert Opin Investig Drugs , 19 :161 , 2010 |
| Abstract : Bui_2010_Expert.Opin.Investig.Drugs_19_161 |
| ESTHER : Bui_2010_Expert.Opin.Investig.Drugs_19_161 |
| PubMedSearch : Bui_2010_Expert.Opin.Investig.Drugs_19_161 |
| PubMedID: 20001561 |
| Title : Lipoprotein-associated phospholipase A(2) and atherosclerosis - Wilensky_2009_Curr.Opin.Lipidol_20_415 |
| Author(s) : Wilensky RL , Macphee CH |
| Ref : Curr Opin Lipidol , 20 :415 , 2009 |
| Abstract : Wilensky_2009_Curr.Opin.Lipidol_20_415 |
| ESTHER : Wilensky_2009_Curr.Opin.Lipidol_20_415 |
| PubMedSearch : Wilensky_2009_Curr.Opin.Lipidol_20_415 |
| PubMedID: 19667981 |
| Title : Darapladib, a reversible lipoprotein-associated phospholipase A2 inhibitor, for the oral treatment of atherosclerosis and coronary artery disease - Riley_2009_IDrugs_12_648 |
| Author(s) : Riley RF , Corson MA |
| Ref : IDrugs , 12 :648 , 2009 |
| Abstract : Riley_2009_IDrugs_12_648 |
| ESTHER : Riley_2009_IDrugs_12_648 |
| PubMedSearch : Riley_2009_IDrugs_12_648 |
| PubMedID: 19790016 |
| Title : The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study - Mohler_2008_J.Am.Coll.Cardiol_51_1632 |
| Author(s) : Mohler ER, 3rd , Ballantyne CM , Davidson MH , Hanefeld M , Ruilope LM , Johnson JL , Zalewski A |
| Ref : J Am Coll Cardiol , 51 :1632 , 2008 |
| Abstract : Mohler_2008_J.Am.Coll.Cardiol_51_1632 |
| ESTHER : Mohler_2008_J.Am.Coll.Cardiol_51_1632 |
| PubMedSearch : Mohler_2008_J.Am.Coll.Cardiol_51_1632 |
| PubMedID: 18436114 |
| Title : Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque - Serruys_2008_Circulation_118_1172 |
| Author(s) : Serruys PW , Garcia-Garcia HM , Buszman P , Erne P , Verheye S , Aschermann M , Duckers H , Bleie O , Dudek D , Botker HE , von Birgelen C , D'Amico D , Hutchinson T , Zambanini A , Mastik F , van Es GA , van der Steen AF , Vince DG , Ganz P , Hamm CW , Wijns W , Zalewski A |
| Ref : Circulation , 118 :1172 , 2008 |
| Abstract : Serruys_2008_Circulation_118_1172 |
| ESTHER : Serruys_2008_Circulation_118_1172 |
| PubMedSearch : Serruys_2008_Circulation_118_1172 |
| PubMedID: 18765397 |
| Title : The identification of clinical candidate SB-480848: a potent inhibitor of lipoprotein-associated phospholipase A2 - Blackie_2003_Bioorg.Med.Chem.Lett_13_1067 |
| Author(s) : Blackie JA , Bloomer JC , Brown MJ , Cheng HY , Hammond B , Hickey DM , Ife RJ , Leach CA , Lewis VA , Macphee CH , Milliner KJ , Moores KE , Pinto IL , Smith SA , Stansfield IG , Stanway SJ , Taylor MA , Theobald CJ |
| Ref : Bioorganic & Medicinal Chemistry Lett , 13 :1067 , 2003 |
| Abstract : Blackie_2003_Bioorg.Med.Chem.Lett_13_1067 |
| ESTHER : Blackie_2003_Bioorg.Med.Chem.Lett_13_1067 |
| PubMedSearch : Blackie_2003_Bioorg.Med.Chem.Lett_13_1067 |
| PubMedID: 12643913 |