Chymostatin
Chymostatin is an oligopeptide produced by various bacteria. Chymostatin is a strong inhibitor of many proteases (mainly not alpha/beta hydrolases), including chymotrypsin, papain, chymotrypsin-like serine proteinases, chymases, and lysosomal cysteine proteinases such as cathepsins A
General
Type : Peptide
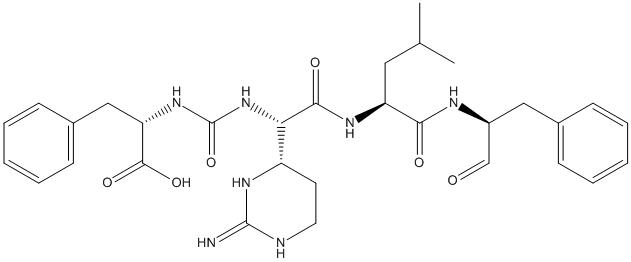
Chemical_Nomenclature : (2S)-2-[[(1S)-1-(2-amino-1,4,5,6-tetrahydropyrimidin-6-yl)-2-[[(2S)-4-methyl-1-oxo-1-[[(2S)-1-oxo-3-phenylpropan-2-yl]amino]pentan-2-yl]amino]-2-oxoethyl]carbamoylamino]-3-phenylpropanoic acid
Canonical SMILES : CC(C)CC(C(=O)NC(CC1=CC=CC=C1)C=O)NC(=O)C(C2CCN=C(N2)N)NC(=O)NC(CC3=CC=CC=C3)C(=O)O
InChI : InChI=1S\/C31H41N7O6\/c1-19(2)15-24(27(40)34-22(18-39)16-20-9-5-3-6-10-20)35-28(41)26(23-13-14-33-30(32)36-23)38-31(44)37-25(29(42)43)17-21-11-7-4-8-12-21\/h3-12,18-19,22-26H,13-17H2,1-2H3,(H,34,40)(H,35,41)(H,42,43)(H3,32,33,36)(H2,37,38,44)\/t22-,23?,24-,25-,26-\/m0\/s1
InChIKey : MRXDGVXSWIXTQL-HYHFHBMOSA-N
Other name(s) : CHEMBL247767,AC1L9E22,C11308,PHE,PHA,LEU,CSI,PRD_000558,SCHEMBL8259564
MW : 607.70
Formula : C31H41N7O6
CAS_number : 9076-44-2
PubChem : 443119
UniChem : MRXDGVXSWIXTQL-HYHFHBMOSA-N
IUPHAR :
Wikipedia :
Target
Families : Chymostatin ligand of proteins in family: Carboxypeptidase_S10
Stucture : 1BCS Wheat Serine carboxypeptidase II + microbial peptide aldehyde inhibitor, chymostatin and arginine at 100 degrees Kelvin
Protein : wheat-cbp02
References (2)
| Title : Comparative analysis of binding energy of chymostatin with human cathepsin A and its homologous proteins by molecular orbital calculation - Yoshida_2006_J.Chem.Inf.Model_46_2093 |
| Author(s) : Yoshida T , Lepp Z , Kadota Y , Satoh Y , Itoh K , Chuman H |
| Ref : J Chem Inf Model , 46 :2093 , 2006 |
| Abstract : Yoshida_2006_J.Chem.Inf.Model_46_2093 |
| ESTHER : Yoshida_2006_J.Chem.Inf.Model_46_2093 |
| PubMedSearch : Yoshida_2006_J.Chem.Inf.Model_46_2093 |
| PubMedID: 16995740 |
| Title : Peptide aldehyde complexes with wheat serine carboxypeptidase II: implications for the catalytic mechanism and substrate specificity - Bullock_1996_J.Mol.Biol_255_714 |
| Author(s) : Bullock TL , Breddam K , Remington SJ |
| Ref : Journal of Molecular Biology , 255 :714 , 1996 |
| Abstract : Bullock_1996_J.Mol.Biol_255_714 |
| ESTHER : Bullock_1996_J.Mol.Biol_255_714 |
| PubMedSearch : Bullock_1996_J.Mol.Biol_255_714 |
| PubMedID: 8636973 |
| Gene_locus related to this paper: wheat-cbp02 |