Benextramine
alpha 2-adrenergic receptors irreversible antagonist. Antagonize several receptors, including nicotinic receptors (nAChRs), muscarinic receptors (mAChRs), neuropeptide Y receptors,and acetylcholinesterase (AChE)
General
Type : Muscarinic antagonist,Not A\/B H target,Adrenergic-Receptor-ligand,Sulfur Compound
Chemical_Nomenclature : N'-[(2-methoxyphenyl)methyl]-N-[2-[2-[6-[(2-methoxyphenyl)methylamino]hexylamino]ethyldisulfanyl]ethyl]hexane-1,6-diamine
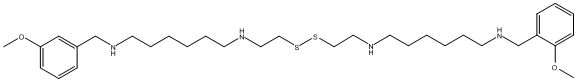
Canonical SMILES : COC1=CC=CC=C1CNCCCCCCNCCSSCCNCCCCCCNCC2=CC=CC=C2OC
InChI : InChI=1S\/C32H54N4O2S2\/c1-37-31-17-9-7-15-29(31)27-35-21-13-5-3-11-19-33-23-25-39-40-26-24-34-20-12-4-6-14-22-36-28-30-16-8-10-18-32(30)38-2\/h7-10,15-18,33-36H,3-6,11-14,19-28H2,1-2H3
InChIKey : IIWOUNLDWKZMQI-UHFFFAOYSA-N
Other name(s) : UNII-2UGY4ETX3L,2UGY4ETX3L,CHEMBL19060
MW : 590.9
Formula : C32H54N4O2S2
CAS_number : 69790-18-7
PubChem : 2317
UniChem : IIWOUNLDWKZMQI-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (10)
| Title : Polymethylene tetraamine backbone as template for the development of biologically active polyamines - Melchiorre_2003_Med.Res.Rev_23_200 |
| Author(s) : Melchiorre C , Antonello A , Banzi R , Bolognesi ML , Minarini A , Rosini M , Tumiatti V |
| Ref : Med Res Rev , 23 :200 , 2003 |
| Abstract : Melchiorre_2003_Med.Res.Rev_23_200 |
| ESTHER : Melchiorre_2003_Med.Res.Rev_23_200 |
| PubMedSearch : Melchiorre_2003_Med.Res.Rev_23_200 |
| PubMedID: 12500289 |
| Title : Design, synthesis, and biological evaluation of symmetrically and unsymmetrically substituted methoctramine-related polyamines as muscular nicotinic receptor noncompetitive antagonists - Rosini_1999_J.Med.Chem_42_5212 |
| Author(s) : Rosini M , Budriesi R , Bixel MG , Bolognesi ML , Chiarini A , Hucho F , Krogsgaard-Larsen P , Mellor IR , Minarini A , Tumiatti V , Usherwood PN , Melchiorre C |
| Ref : Journal of Medicinal Chemistry , 42 :5212 , 1999 |
| Abstract : Rosini_1999_J.Med.Chem_42_5212 |
| ESTHER : Rosini_1999_J.Med.Chem_42_5212 |
| PubMedSearch : Rosini_1999_J.Med.Chem_42_5212 |
| PubMedID: 10602706 |
| Title : Acetylcholinesterase Noncovalent Inhibitors Based on a Polyamine Backbone for Potential Use against Alzheimer's Disease - |
| Author(s) : Melchiorre C , Andrisano V , Bolognesi ML , Budriesi R , Cavalli A , Cavrini V , Rosini M , Tumiatti V , Recanatini M |
| Ref : Journal of Medicinal Chemistry , 41 :4186 , 1998 |
| PubMedID: 9784091 |
| Title : Universal template approach to drug design: polyamines as selective muscarinic receptor antagonists - Bolognesi_1998_J.Med.Chem_41_4150 |
| Author(s) : Bolognesi ML , Minarini A , Budriesi R , Cacciaguerra S , Chiarini A , Spampinato S , Tumiatti V , Melchiorre C |
| Ref : Journal of Medicinal Chemistry , 41 :4150 , 1998 |
| Abstract : Bolognesi_1998_J.Med.Chem_41_4150 |
| ESTHER : Bolognesi_1998_J.Med.Chem_41_4150 |
| PubMedSearch : Bolognesi_1998_J.Med.Chem_41_4150 |
| PubMedID: 9767650 |
| Title : Discrimination by benextramine between the NPY-Y1 receptor subtypes present in rabbit isolated vas deferens and saphenous vein - Palea_1995_Br.J.Pharmacol_115_3 |
| Author(s) : Palea S , Corsi M , Rimland JM , Trist DG |
| Ref : British Journal of Pharmacology , 115 :3 , 1995 |
| Abstract : Palea_1995_Br.J.Pharmacol_115_3 |
| ESTHER : Palea_1995_Br.J.Pharmacol_115_3 |
| PubMedSearch : Palea_1995_Br.J.Pharmacol_115_3 |
| PubMedID: 7647980 |
| Title : Failure of the putative neuropeptide Y antagonists, benextramine and PYX-2, to inhibit Y2 receptors in rat isolated prostatic vas deferens - Palea_1995_Br.J.Pharmacol_116_2401 |
| Author(s) : Palea S , Corsi M , Rimland JM , Trist DG , Ratti E |
| Ref : British Journal of Pharmacology , 116 :2401 , 1995 |
| Abstract : Palea_1995_Br.J.Pharmacol_116_2401 |
| ESTHER : Palea_1995_Br.J.Pharmacol_116_2401 |
| PubMedSearch : Palea_1995_Br.J.Pharmacol_116_2401 |
| PubMedID: 8581275 |
| Title : Binding profile of benextramine at neuropeptide Y receptor subtypes in rat brain areas - Melchiorre_1994_Eur.J.Pharmacol_265_93 |
| Author(s) : Melchiorre C , Romualdi P , Bolognesi ML , Donatini A , Ferri S |
| Ref : European Journal of Pharmacology , 265 :93 , 1994 |
| Abstract : Melchiorre_1994_Eur.J.Pharmacol_265_93 |
| ESTHER : Melchiorre_1994_Eur.J.Pharmacol_265_93 |
| PubMedSearch : Melchiorre_1994_Eur.J.Pharmacol_265_93 |
| PubMedID: 7883034 |
| Title : Polymethylene tetraamines: a novel class of cardioselective M2-antagonists - |
| Author(s) : Melchiorre C |
| Ref : Med Res Rev , 10 :327 , 1990 |
| PubMedID: 2196404 |
| Title : Polymethylene tetraamines as muscarinic receptor probes - Melchiorre_1989_Trends.Pharmacol.Sci_Suppl_55 |
| Author(s) : Melchiorre C , Minarini A , Angeli P , Giardina D , Gulini U , Quaglia W |
| Ref : Trends in Pharmacological Sciences , Suppl :55 , 1989 |
| Abstract : Melchiorre_1989_Trends.Pharmacol.Sci_Suppl_55 |
| ESTHER : Melchiorre_1989_Trends.Pharmacol.Sci_Suppl_55 |
| PubMedSearch : Melchiorre_1989_Trends.Pharmacol.Sci_Suppl_55 |
| PubMedID: 2694524 |
| Title : Potentiation and inhibition of nicotinic effects on striated muscle by the tetramine disulfide benextramine - Benfey_1980_Can.J.Physiol.Pharmacol_58_984 |
| Author(s) : Benfey BG , Brasili L , Melchiorre C , Belleau B |
| Ref : Canadian Journal of Physiology & Pharmacology , 58 :984 , 1980 |
| Abstract : Benfey_1980_Can.J.Physiol.Pharmacol_58_984 |
| ESTHER : Benfey_1980_Can.J.Physiol.Pharmacol_58_984 |
| PubMedSearch : Benfey_1980_Can.J.Physiol.Pharmacol_58_984 |
| PubMedID: 6112056 |