Aprophen
Anticholinergic agent, anti muscarinic. Not really an inhibitor of cholinesterases but interaction of aprophen with cholinesterase protect from inhibition by OP. Both inhibitor and substrate of BChE . Used to make binary compounds with antimuscarinic profile and protection effect on cholinesterases . Compounds (carbamyl substitution on the phenyl ring called carbaphen)
General
Type : Muscarinic antagonist,Propionate
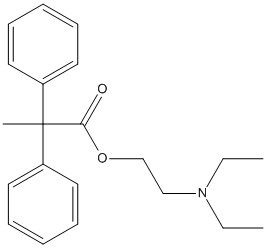
Chemical_Nomenclature : 2-diethylaminoethyl 2,2-diphenylpropanoate \; alpha-Methyl-alpha-phenylbenzeeacetic acid 2-(diethylamino)ethyl ester
Canonical SMILES : CCN(CC)CCOC(=O)C(C)(C1=CC=CC=C1)C2=CC=CC=C2
InChI : InChI=1S\/C21H27NO2\/c1-4-22(5-2)16-17-24-20(23)21(3,18-12-8-6-9-13-18)19-14-10-7-11-15-19\/h6-15H,4-5,16-17H2,1-3H3
InChIKey : DIDYGLSKVUKRRP-UHFFFAOYSA-N
Other name(s) : Aprofen,Aprofene,Aprophenum
MW : 325.445
Formula : C21H27NO2
CAS_number : 3563-01-7
PubChem : 71128
UniChem : DIDYGLSKVUKRRP-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (46)
| Title : [The pharmacokinetic interaction of cholinolytics and cholinesterase reactivators as a reflection of the modulation of their binding in blood plasma and in brain tissue] - Elaeva_1997_Eksp.Klin.Farmakol_60_64 |
| Author(s) : Elaeva NL , Predtechenskii MB , Kul'bitskii GN , Babaina EV , Trefilov VV |
| Ref : Eksperimentalnaia i Klinicheskaia Farmakologiia , 60 :64 , 1997 |
| Abstract : Elaeva_1997_Eksp.Klin.Farmakol_60_64 |
| ESTHER : Elaeva_1997_Eksp.Klin.Farmakol_60_64 |
| PubMedSearch : Elaeva_1997_Eksp.Klin.Farmakol_60_64 |
| PubMedID: 9376763 |
| Title : Differential effects of anticholinergic drugs on paired discrimination performance - Grauer_1996_Pharmacol.Biochem.Behav_53_463 |
| Author(s) : Grauer E , Kapon J |
| Ref : Pharmacol Biochem Behav , 53 :463 , 1996 |
| Abstract : Grauer_1996_Pharmacol.Biochem.Behav_53_463 |
| ESTHER : Grauer_1996_Pharmacol.Biochem.Behav_53_463 |
| PubMedSearch : Grauer_1996_Pharmacol.Biochem.Behav_53_463 |
| PubMedID: 8808159 |
| Title : A study of the N-methyl-D-aspartate antagonistic properties of anticholinergic drugs - McDonough_1995_Pharmacol.Biochem.Behav_51_249 |
| Author(s) : McDonough JH, Jr. , Shih TM |
| Ref : Pharmacol Biochem Behav , 51 :249 , 1995 |
| Abstract : McDonough_1995_Pharmacol.Biochem.Behav_51_249 |
| ESTHER : McDonough_1995_Pharmacol.Biochem.Behav_51_249 |
| PubMedSearch : McDonough_1995_Pharmacol.Biochem.Behav_51_249 |
| PubMedID: 7667336 |
| Title : Synthesis and antimuscarinic activity of 2-[N-(ethyl)-(N-beta- hydroxyethyl)]aminoethyl 2,2-diphenylpropionate, a metabolite of aprophen - Brown_1993_J.Pharm.Sci_82_563 |
| Author(s) : Brown ND , Leader H , Phillips LR , Smejkal RM , Gordon RK , Chiang PK |
| Ref : J Pharm Sci , 82 :563 , 1993 |
| Abstract : Brown_1993_J.Pharm.Sci_82_563 |
| ESTHER : Brown_1993_J.Pharm.Sci_82_563 |
| PubMedSearch : Brown_1993_J.Pharm.Sci_82_563 |
| PubMedID: 8331525 |
| Title : Relationship of three-dimensional structure of muscarinic antagonists to antimuscarinic activity: structure of thiodeacylaprophen hydrochloride - Karle_1992_Acta.Crystallogr.B_48_208 |
| Author(s) : Karle JM , Karle IL , Gordon RK , Chiang PK |
| Ref : Acta Crystallographica B , 48 :208 , 1992 |
| Abstract : Karle_1992_Acta.Crystallogr.B_48_208 |
| ESTHER : Karle_1992_Acta.Crystallogr.B_48_208 |
| PubMedSearch : Karle_1992_Acta.Crystallogr.B_48_208 |
| PubMedID: 1515109 |
| Title : Muscarinic receptor subtype specificity of (N,N-dialkylamino)alkyl 2- cyclohexyl-2-phenylpropionates: cylexphenes (cyclohexyl-substituted aprophen analogues) - Leader_1992_J.Med.Chem_35_1290 |
| Author(s) : Leader H , Gordon RK , Baumgold J , Boyd VL , Newman AH , Smejkal RM , Chiang PK |
| Ref : Journal of Medicinal Chemistry , 35 :1290 , 1992 |
| Abstract : Leader_1992_J.Med.Chem_35_1290 |
| ESTHER : Leader_1992_J.Med.Chem_35_1290 |
| PubMedSearch : Leader_1992_J.Med.Chem_35_1290 |
| PubMedID: 1560441 |
| Title : Successful pretreatment\/therapy of soman, sarin and VX intoxication - Lennox_1992_Drug.Chem.Toxicol_15_271 |
| Author(s) : Lennox WJ , Harris LW , Anderson DR , Solana RP , Murrow ML , Wade JV |
| Ref : Drug & Chemical Toxicology , 15 :271 , 1992 |
| Abstract : Lennox_1992_Drug.Chem.Toxicol_15_271 |
| ESTHER : Lennox_1992_Drug.Chem.Toxicol_15_271 |
| PubMedSearch : Lennox_1992_Drug.Chem.Toxicol_15_271 |
| PubMedID: 1459040 |
| Title : M1 muscarinic antagonists interact with sigma recognition sites [published erratum appears in Life Sci 1992\;50(3):245] - Hudkins_1991_Life.Sci_49_1229 |
| Author(s) : Hudkins RL , DeHaven-Hudkins DL |
| Ref : Life Sciences , 49 :1229 , 1991 |
| Abstract : Hudkins_1991_Life.Sci_49_1229 |
| ESTHER : Hudkins_1991_Life.Sci_49_1229 |
| PubMedSearch : Hudkins_1991_Life.Sci_49_1229 |
| PubMedID: 1658507 |
| Title : Anticholinergics: effects on thermoregulation and performance in rats - Matthew_1991_Neurosci.Biobehav.Rev_15_141 |
| Author(s) : Matthew CB |
| Ref : Neurosci Biobehav Rev , 15 :141 , 1991 |
| Abstract : Matthew_1991_Neurosci.Biobehav.Rev_15_141 |
| ESTHER : Matthew_1991_Neurosci.Biobehav.Rev_15_141 |
| PubMedSearch : Matthew_1991_Neurosci.Biobehav.Rev_15_141 |
| PubMedID: 2052187 |
| Title : Anticonvulsants for poisoning by the organophosphorus compound soman: pharmacological mechanisms - Shih_1991_Neurosci.Biobehav.Rev_15_349 |
| Author(s) : Shih TM , Koviak TA , Capacio BR |
| Ref : Neurosci Biobehav Rev , 15 :349 , 1991 |
| Abstract : Shih_1991_Neurosci.Biobehav.Rev_15_349 |
| ESTHER : Shih_1991_Neurosci.Biobehav.Rev_15_349 |
| PubMedSearch : Shih_1991_Neurosci.Biobehav.Rev_15_349 |
| PubMedID: 1683477 |
| Title : Effects of some antimuscarinics alone and in combination with chlordiazepoxide on punished and nonpunished behavior of rats - Witkin_1991_Pharmacol.Biochem.Behav_39_453 |
| Author(s) : Witkin JM , Witkin KM |
| Ref : Pharmacol Biochem Behav , 39 :453 , 1991 |
| Abstract : Witkin_1991_Pharmacol.Biochem.Behav_39_453 |
| ESTHER : Witkin_1991_Pharmacol.Biochem.Behav_39_453 |
| PubMedSearch : Witkin_1991_Pharmacol.Biochem.Behav_39_453 |
| PubMedID: 1946585 |
| Title : Isolation and identification of beta-hydroxyethylaprophen: a urinary metabolite of aprophen in rats - Brown_1991_J.Chromatogr_563_466 |
| Author(s) : Brown ND , Phillips LR , Leader H , Chiang PK |
| Ref : Journal of Chromatography , 563 :466 , 1991 |
| Abstract : Brown_1991_J.Chromatogr_563_466 |
| ESTHER : Brown_1991_J.Chromatogr_563_466 |
| PubMedSearch : Brown_1991_J.Chromatogr_563_466 |
| PubMedID: 2056011 |
| Title : Anticonvulsant actions of anticholinergic drugs in soman poisoning - Capacio_1991_Epilepsia_32_604 |
| Author(s) : Capacio BR , Shih TM |
| Ref : Epilepsia , 32 :604 , 1991 |
| Abstract : Capacio_1991_Epilepsia_32_604 |
| ESTHER : Capacio_1991_Epilepsia_32_604 |
| PubMedSearch : Capacio_1991_Epilepsia_32_604 |
| PubMedID: 1915166 |
| Title : Physostigmine (alone and together with adjunct) pretreatment against soman, sarin, tabun and VX intoxication - Harris_1991_Drug.Chem.Toxicol_14_265 |
| Author(s) : Harris LW , Talbot BG , Lennox WJ , Anderson DR , Solana RP |
| Ref : Drug & Chemical Toxicology , 14 :265 , 1991 |
| Abstract : Harris_1991_Drug.Chem.Toxicol_14_265 |
| ESTHER : Harris_1991_Drug.Chem.Toxicol_14_265 |
| PubMedSearch : Harris_1991_Drug.Chem.Toxicol_14_265 |
| PubMedID: 1935706 |
| Title : Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. - Lockridge_1990_Pharmacol.Ther_47_35 |
| Author(s) : Lockridge O |
| Ref : Pharmacol Ther , 47 :35 , 1990 |
| Abstract : Lockridge_1990_Pharmacol.Ther_47_35 |
| ESTHER : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedSearch : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedID: 2195556 |
| Gene_locus related to this paper: human-BCHE |
| Title : Structural comparison of the potent antimuscarinic agent azaprophen hydrochloride with aprophen hydrochloride and structurally related antimuscarinic agents - Karle_1990_Acta.Crystallogr.B_46_215 |
| Author(s) : Karle JM , Karle IL , Chiang PK |
| Ref : Acta Crystallographica B , 46 :215 , 1990 |
| Abstract : Karle_1990_Acta.Crystallogr.B_46_215 |
| ESTHER : Karle_1990_Acta.Crystallogr.B_46_215 |
| PubMedSearch : Karle_1990_Acta.Crystallogr.B_46_215 |
| PubMedID: 2344395 |
| Title : Relationship of the behavioral effects of aprophen, atropine and scopolamine to antagonism of the behavioral effects of physostigmine - Genovese_1990_Pharmacol.Biochem.Behav_37_117 |
| Author(s) : Genovese RF , Elsmore TF , Witkin JM |
| Ref : Pharmacol Biochem Behav , 37 :117 , 1990 |
| Abstract : Genovese_1990_Pharmacol.Biochem.Behav_37_117 |
| ESTHER : Genovese_1990_Pharmacol.Biochem.Behav_37_117 |
| PubMedSearch : Genovese_1990_Pharmacol.Biochem.Behav_37_117 |
| PubMedID: 2263653 |
| Title : Aprophit: an irreversible antagonist for muscarinic receptors - Newman_1990_Biochem.Pharmacol_40_1357 |
| Author(s) : Newman AH , Covington J , Oleshansky M , Jackson BW , Weissman BA , Leader H , Chiang PK |
| Ref : Biochemical Pharmacology , 40 :1357 , 1990 |
| Abstract : Newman_1990_Biochem.Pharmacol_40_1357 |
| ESTHER : Newman_1990_Biochem.Pharmacol_40_1357 |
| PubMedSearch : Newman_1990_Biochem.Pharmacol_40_1357 |
| PubMedID: 2403389 |
| Title : [Delivery in prolonged pregnancy following preparation and induction with aprofen and prednisolone] - Grudev_1990_Akush.Ginekol.(Sofiia)_29_14 |
| Author(s) : Grudev D |
| Ref : Akush Ginekol (Sofiia) , 29 :14 , 1990 |
| Abstract : Grudev_1990_Akush.Ginekol.(Sofiia)_29_14 |
| ESTHER : Grudev_1990_Akush.Ginekol.(Sofiia)_29_14 |
| PubMedSearch : Grudev_1990_Akush.Ginekol.(Sofiia)_29_14 |
| PubMedID: 2252139 |
| Title : Binary antidotes for organophosphate poisoning: aprophen analogues that are both antimuscarinics and carbamates - Leader_1989_J.Med.Chem_32_1522 |
| Author(s) : Leader H , Smejkal RM , Payne CS , Padilla FN , Doctor BP , Gordon RK , Chiang PK |
| Ref : Journal of Medicinal Chemistry , 32 :1522 , 1989 |
| Abstract : Leader_1989_J.Med.Chem_32_1522 |
| ESTHER : Leader_1989_J.Med.Chem_32_1522 |
| PubMedSearch : Leader_1989_J.Med.Chem_32_1522 |
| PubMedID: 2738887 |
| Title : Side effects of therapeutic drugs against organophosphate poisoning - Wolthuis_1989_Neurotoxicol.Teratol_11_221 |
| Author(s) : Wolthuis OL , Philippens IH , Vanwersch RA |
| Ref : Neurotoxicology & Teratology , 11 :221 , 1989 |
| Abstract : Wolthuis_1989_Neurotoxicol.Teratol_11_221 |
| ESTHER : Wolthuis_1989_Neurotoxicol.Teratol_11_221 |
| PubMedSearch : Wolthuis_1989_Neurotoxicol.Teratol_11_221 |
| PubMedID: 2666846 |
| Title : [The results of inducing labor with aprofen] - |
| Author(s) : Khadzhiev A , Dimitrova V , Filipov E |
| Ref : Akush Ginekol (Sofiia) , 27 :38 , 1988 |
| PubMedID: 3245549 |
| Title : Reversal of inhibition of prolactin secretion in cultured pituitary cells by muscarinic antagonists - Beach_1988_J.Pharmacol.Exp.Ther_246_548 |
| Author(s) : Beach JE , Smallridge RC , Chiang PK , Fein HG |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 246 :548 , 1988 |
| Abstract : Beach_1988_J.Pharmacol.Exp.Ther_246_548 |
| ESTHER : Beach_1988_J.Pharmacol.Exp.Ther_246_548 |
| PubMedSearch : Beach_1988_J.Pharmacol.Exp.Ther_246_548 |
| PubMedID: 3404446 |
| Title : Effects of muscarinic pharmacophores on the cholinergic regulation of catecholamine secretion from perfused adrenal glands - Nakazato_1988_Arch.Int.Pharmacodyn.Ther_293_209 |
| Author(s) : Nakazato Y , Oleshansky MA , Chiang PK |
| Ref : Archives Internationales de Pharmacodynamie et de Therapie , 293 :209 , 1988 |
| Abstract : Nakazato_1988_Arch.Int.Pharmacodyn.Ther_293_209 |
| ESTHER : Nakazato_1988_Arch.Int.Pharmacodyn.Ther_293_209 |
| PubMedSearch : Nakazato_1988_Arch.Int.Pharmacodyn.Ther_293_209 |
| PubMedID: 3421778 |
| Title : Desethylaprophen: a metabolite of aprophen with antimuscarinic activities - Brown_1988_J.Pharm.Sci_77_145 |
| Author(s) : Brown ND , Smejkal RM , Breuer E , Doctor BP , Chiang PK |
| Ref : J Pharm Sci , 77 :145 , 1988 |
| Abstract : Brown_1988_J.Pharm.Sci_77_145 |
| ESTHER : Brown_1988_J.Pharm.Sci_77_145 |
| PubMedSearch : Brown_1988_J.Pharm.Sci_77_145 |
| PubMedID: 3258910 |
| Title : Effects of selected muscarinic cholinergic antagonists on [3H]acetylcholine release from rat hippocampal slices - Pohorecki_1988_J.Pharmacol.Exp.Ther_244_213 |
| Author(s) : Pohorecki R , Head R , Domino EF |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 244 :213 , 1988 |
| Abstract : Pohorecki_1988_J.Pharmacol.Exp.Ther_244_213 |
| ESTHER : Pohorecki_1988_J.Pharmacol.Exp.Ther_244_213 |
| PubMedSearch : Pohorecki_1988_J.Pharmacol.Exp.Ther_244_213 |
| PubMedID: 3335998 |
| Title : [Use of aprophen in the preparation for and induction of labor] - |
| Author(s) : Grudev D , Novachkov L , Geshev G , Krushkov I |
| Ref : Akush Ginekol , 27 :39 , 1988 |
| PubMedID: 3218679 |
| Title : Structure-binding relationship of quinuclidinyl benzilate analogs on N4TG1 neuroblastoma muscarinic receptors - Chang_1988_Neurochem.Res_13_455 |
| Author(s) : Chang YF |
| Ref : Neurochem Res , 13 :455 , 1988 |
| Abstract : Chang_1988_Neurochem.Res_13_455 |
| ESTHER : Chang_1988_Neurochem.Res_13_455 |
| PubMedSearch : Chang_1988_Neurochem.Res_13_455 |
| PubMedID: 3405371 |
| Title : The muscarinic antagonists aprophen and benactyzine are noncompetitive inhibitors of the nicotinic acetylcholine receptor - Amitai_1987_Mol.Pharmacol_32_678 |
| Author(s) : Amitai G , Herz JM , Bruckstein R , Luz-Chapman S |
| Ref : Molecular Pharmacology , 32 :678 , 1987 |
| Abstract : Amitai_1987_Mol.Pharmacol_32_678 |
| ESTHER : Amitai_1987_Mol.Pharmacol_32_678 |
| PubMedSearch : Amitai_1987_Mol.Pharmacol_32_678 |
| PubMedID: 3683366 |
| Title : Comparison of in vitro actions with behavioral effects of antimuscarinic agents - Witkin_1987_J.Pharmacol.Exp.Ther_242_796 |
| Author(s) : Witkin JM , Gordon RK , Chiang PK |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 242 :796 , 1987 |
| Abstract : Witkin_1987_J.Pharmacol.Exp.Ther_242_796 |
| ESTHER : Witkin_1987_J.Pharmacol.Exp.Ther_242_796 |
| PubMedSearch : Witkin_1987_J.Pharmacol.Exp.Ther_242_796 |
| PubMedID: 3498817 |
| Title : Kinetic investigations into the interactions of aprophen with cholinesterases and a carboxylesterase - Rush_1986_Biochem.Pharmacol_35_4167 |
| Author(s) : Rush RS , Doctor BP , Wolfe AD |
| Ref : Biochemical Pharmacology , 35 :4167 , 1986 |
| Abstract : Rush_1986_Biochem.Pharmacol_35_4167 |
| ESTHER : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedSearch : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedID: 3098245 |
| Title : Enzymatic synthesis of a false cholinergic neurotransmitter: diethylaminoethyl acetate as a muscarinic agonist analog of acetylcholine - Miura_1986_Gen.Pharmacol_17_477 |
| Author(s) : Miura GA , Chiang PK , Gordon RK , Doctor BP , Twine CE , Philip A , Kepler JA , Carroll FI |
| Ref : General Pharmacology , 17 :477 , 1986 |
| Abstract : Miura_1986_Gen.Pharmacol_17_477 |
| ESTHER : Miura_1986_Gen.Pharmacol_17_477 |
| PubMedSearch : Miura_1986_Gen.Pharmacol_17_477 |
| PubMedID: 2875921 |
| Title : Disposition of aprophen in rats - Aarbakke_1986_J.Pharm.Pharmacol_38_928 |
| Author(s) : Aarbakke J , Miura GA , Brown ND , Gray RR , Gordon RK , Doctor BP , Chiang PK |
| Ref : J Pharm Pharmacol , 38 :928 , 1986 |
| Abstract : Aarbakke_1986_J.Pharm.Pharmacol_38_928 |
| ESTHER : Aarbakke_1986_J.Pharm.Pharmacol_38_928 |
| PubMedSearch : Aarbakke_1986_J.Pharm.Pharmacol_38_928 |
| PubMedID: 2880971 |
| Title : Aprophen: a substrate and inhibitor of butyrylcholinesterase and carboxylesterases - Rush_1985_Biochem.Pharmacol_34_2063 |
| Author(s) : Rush RS , Ralston JS , Wolfe AD |
| Ref : Biochemical Pharmacology , 34 :2063 , 1985 |
| Abstract : Rush_1985_Biochem.Pharmacol_34_2063 |
| ESTHER : Rush_1985_Biochem.Pharmacol_34_2063 |
| PubMedSearch : Rush_1985_Biochem.Pharmacol_34_2063 |
| PubMedID: 4004924 |
| Title : Comparative effects of aprophen, atropine and benactyzine on central and peripheral cholinoceptors and on acetylcholinesterase - |
| Author(s) : Dawson RM , Freeman SE , Paddle BM |
| Ref : Biochemical Pharmacology , 34 :1577 , 1985 |
| PubMedID: 3994766 |
| Title : Treatment of poisoning by soman - Leadbeater_1985_Fundam.Appl.Toxicol_5_S225 |
| Author(s) : Leadbeater L , Inns RH , Rylands JM |
| Ref : Fundamental & Applied Toxicology , 5 :S225 , 1985 |
| Abstract : Leadbeater_1985_Fundam.Appl.Toxicol_5_S225 |
| ESTHER : Leadbeater_1985_Fundam.Appl.Toxicol_5_S225 |
| PubMedSearch : Leadbeater_1985_Fundam.Appl.Toxicol_5_S225 |
| PubMedID: 4092890 |
| Title : [Participation of cholinergic mechanisms in realizing the effects of bicuculline] - Ostrovskaia_1978_Biull.Eksp.Biol.Med_86_192 |
| Author(s) : Ostrovskaia RU , Molodavkin GM |
| Ref : Biulleten Eksperimentalnoi Biologii i Meditsiny , 86 :192 , 1978 |
| Abstract : Ostrovskaia_1978_Biull.Eksp.Biol.Med_86_192 |
| ESTHER : Ostrovskaia_1978_Biull.Eksp.Biol.Med_86_192 |
| PubMedSearch : Ostrovskaia_1978_Biull.Eksp.Biol.Med_86_192 |
| PubMedID: 210864 |
| Title : [The determination of aprophen in medicinal forms] - |
| Author(s) : Uspenskaia SI |
| Ref : Farmatsiia , 22 :58 , 1973 |
| PubMedID: 4710866 |
| Title : [Microcrystalloscopic reactions on aprophen, tiphen, spasmolytin and euphylline and their use in the analysis of drug forms] - |
| Author(s) : Pozdniakova VT , Chaplinskaia MG , Ushbaev KU |
| Ref : Farm Zh , 24 :37 , 1969 |
| PubMedID: 4389432 |
| Title : [Study of self-sterilizing properties of aprophen, tiphen, spasmolytin and diprophene (on technology of injections and solutions)] - |
| Author(s) : Chaplins'ka MG , Gubina MM |
| Ref : Farm Zh , 24 :64 , 1969 |
| PubMedID: 4390330 |
| Title : [Comparative effect of central cholinolytics amisyl, aprophen and corresponding quinuclidine esters on conditioned reflex activity and behavior of rats] - |
| Author(s) : Mashkovskii MD , Roshchina LF |
| Ref : Farmakologiia i Toksikologiia , 32 :16 , 1969 |
| PubMedID: 5770307 |
| Title : [Comparative cholinolytic activity of amizil, aprophen and of their corresponding quinuclidine esters] - |
| Author(s) : Mashkovskii MD , Zaitseva KA |
| Ref : Farmakologiia i Toksikologiia , 30 :36 , 1967 |
| PubMedID: 5594357 |
| Title : [Effect of amizil, aprophen and their corresponding quinuclidine ethers on the bioelectrical activity of the brain] - |
| Author(s) : Mashkovskii MD , Poshchina LF |
| Ref : Farmakologiia i Toksikologiia , 30 :415 , 1967 |
| PubMedID: 5598891 |
| Title : [Study of the stability of aprophen solution for injections] - |
| Author(s) : Chaplinskaia MG , Dolzhanskaia RN |
| Ref : Farmatsiia , 16 :20 , 1967 |
| PubMedID: 5611762 |