AEBSF
serine protease inhibitor
General
Type : Sulfur Compound,Sulfonyl
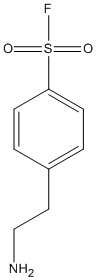
Chemical_Nomenclature : 4-(2-aminoethyl)benzenesulfonyl fluoride
Canonical SMILES : C1=CC(=CC=C1CCN)S(=O)(=O)F
InChI : InChI=1S\/C8H10FNO2S\/c9-13(11,12)8-3-1-7(2-4-8)5-6-10\/h1-4H,5-6,10H2
InChIKey : MGSKVZWGBWPBTF-UHFFFAOYSA-N
Other name(s) : 4-(2-Aminoethyl)benzenesulfonyl fluoride,Pefabloc,4-(2-Aminoethyl)benzenesulfonylfluoride,Benzenesulfonyl fluoride, 4-(2-aminoethyl)-
MW : 203.23
Formula : C8H10FNO2S
CAS_number : 34284-75-8
PubChem : 1701
UniChem : MGSKVZWGBWPBTF-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : AEBSF ligand of proteins in family: S9N_PREPL_Peptidase_S9 || 5_AlphaBeta_hydrolase || DPP4N_Peptidase_S9 || Tiancimycin-TnmK-Tripeptidase-HIP
Stucture :
Protein : pig-dpp4 || leima-OPB || halo1-d0lmj0 || human-FAP || myctu-ym24
References (5)
| Title : 2.1 A crystal structure of the Mycobacterium tuberculosis serine hydrolase, Hip1, in its anhydro-form (Anhydrohip1) - Brooks_2022_Biochem.Biophys.Res.Commun_630_57 |
| Author(s) : Brooks CL , Ostrov DA , Schumann NC , Kakkad S , Li D , Pena K , Williams BP , Goldfarb NE |
| Ref : Biochemical & Biophysical Research Communications , 630 :57 , 2022 |
| Abstract : Brooks_2022_Biochem.Biophys.Res.Commun_630_57 |
| ESTHER : Brooks_2022_Biochem.Biophys.Res.Commun_630_57 |
| PubMedSearch : Brooks_2022_Biochem.Biophys.Res.Commun_630_57 |
| PubMedID: 36148729 |
| Gene_locus related to this paper: myctu-ym24 |
| Title : Polysaccharide utilization loci-driven enzyme discovery reveals BD-FAE: a bifunctional feruloyl and acetyl xylan esterase active on complex natural xylans - Hameleers_2021_Biotechnol.Biofuels_14_127 |
| Author(s) : Hameleers L , Penttinen L , Ikonen M , Jaillot L , Faure R , Terrapon N , Deuss PJ , Hakulinen N , Master ER , Jurak E |
| Ref : Biotechnol Biofuels , 14 :127 , 2021 |
| Abstract : Hameleers_2021_Biotechnol.Biofuels_14_127 |
| ESTHER : Hameleers_2021_Biotechnol.Biofuels_14_127 |
| PubMedSearch : Hameleers_2021_Biotechnol.Biofuels_14_127 |
| PubMedID: 34059129 |
| Gene_locus related to this paper: 9zzzz-6XYC |
| Title : Crystal structure of Leishmania major oligopeptidase B gives insight into the enzymatic properties of a trypanosomatid virulence factor - McLuskey_2010_J.Biol.Chem_285_39249 |
| Author(s) : McLuskey K , Paterson NG , Bland ND , Isaacs NW , Mottram JC |
| Ref : Journal of Biological Chemistry , 285 :39249 , 2010 |
| Abstract : McLuskey_2010_J.Biol.Chem_285_39249 |
| ESTHER : McLuskey_2010_J.Biol.Chem_285_39249 |
| PubMedSearch : McLuskey_2010_J.Biol.Chem_285_39249 |
| PubMedID: 20926390 |
| Gene_locus related to this paper: leima-OPB |
| Title : Structural characterization and reversal of the natural organophosphate resistance of a D-type esterase, Saccharomyces cerevisiae S-formylglutathione hydrolase - Legler_2008_Biochemistry_47_9592 |
| Author(s) : Legler PM , Kumaran D , Swaminathan S , Studier FW , Millard CB |
| Ref : Biochemistry , 47 :9592 , 2008 |
| Abstract : Legler_2008_Biochemistry_47_9592 |
| ESTHER : Legler_2008_Biochemistry_47_9592 |
| PubMedSearch : Legler_2008_Biochemistry_47_9592 |
| PubMedID: 18707125 |
| Gene_locus related to this paper: yeast-yjg8 |
| Title : Rigidity and flexibility of dipeptidyl peptidase IV: crystal structures of and docking experiments with DPIV - Engel_2006_J.Mol.Biol_355_768 |
| Author(s) : Engel M , Hoffmann T , Manhart S , Heiser U , Chambre S , Huber R , Demuth HU , Bode W |
| Ref : Journal of Molecular Biology , 355 :768 , 2006 |
| Abstract : Engel_2006_J.Mol.Biol_355_768 |
| ESTHER : Engel_2006_J.Mol.Biol_355_768 |
| PubMedSearch : Engel_2006_J.Mol.Biol_355_768 |
| PubMedID: 16330047 |
| Gene_locus related to this paper: pig-dpp4 |