Decamethonium
Decamethonium is a short acting depolarizing muscle relaxant or neuromuscular blocking agent, and is used in anesthesia to induce paralysis. It is similar to acetylcholine and acts as a partial agonist of the nicotinic acetylcholine receptor. Skeletal muscle relaxant. Ligand of small conductance Ca2+-activated K+ (SKca) channels
General
Type : Bisquaternary,Trimethylammonium,Neuromuscular Nondepolarizing Agents,Nicotinic antagonist,Drug,Not A\/B H target
Chemical_Nomenclature : 1,10-bis(trimethylammonium)decane
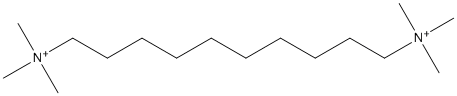
Canonical SMILES : C[N+](C)(C)CCCCCCCCCC[N+](C)(C)C
InChI : InChI=1S\/C16H38N2\/c1-17(2,3)15-13-11-9-7-8-10-12-14-16-18(4,5)6\/h7-16H2,1-6H3\/q+2
InChIKey : MTCUAOILFDZKCO-UHFFFAOYSA-N
Other name(s) : N,N,N,N',N',N'-Hexamethyl-1,10-decanediaminium dibromide,decamethylenebis-[trimethylammonium bromide],C-10,Syncurine,DME
MW : 418.3
Formula : C16H38Br2N2
CAS_number : 541-22-0
PubChem : 2968
UniChem : MTCUAOILFDZKCO-UHFFFAOYSA-N
IUPHAR :
Wikipedia : Decamethonium
Target
Families : Decamethonium ligand of proteins in family: BCHE
Stucture :
Protein : torca-ACHE || mouse-ACHE || human-BCHE
References (15)
| Title : Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study - Rosenberry_2017_Molecules_22_ |
| Author(s) : Rosenberry TL , Brazzolotto X , Macdonald IR , Wandhammer M , Trovaslet-Leroy M , Darvesh S , Nachon F |
| Ref : Molecules , 22 : , 2017 |
| Abstract : Rosenberry_2017_Molecules_22_ |
| ESTHER : Rosenberry_2017_Molecules_22_ |
| PubMedSearch : Rosenberry_2017_Molecules_22_ |
| PubMedID: 29186056 |
| Gene_locus related to this paper: human-BCHE |
| Title : beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies - Bartolini_2003_Biochem.Pharmacol_65_407 |
| Author(s) : Bartolini M , Bertucci C , Cavrini V , Andrisano V |
| Ref : Biochemical Pharmacology , 65 :407 , 2003 |
| Abstract : Bartolini_2003_Biochem.Pharmacol_65_407 |
| ESTHER : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedSearch : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedID: 12527333 |
| Title : Crystal structure of mouse acetylcholinesterase. A peripheral site- occluding loop in a tetrameric assembly - Bourne_1999_J.Biol.Chem_274_2963 |
| Author(s) : Bourne Y , Taylor P , Bougis PE , Marchot P |
| Ref : Journal of Biological Chemistry , 274 :2963 , 1999 |
| Abstract : Bourne_1999_J.Biol.Chem_274_2963 |
| ESTHER : Bourne_1999_J.Biol.Chem_274_2963 |
| PubMedSearch : Bourne_1999_J.Biol.Chem_274_2963 |
| PubMedID: 9915834 |
| Gene_locus related to this paper: mouse-ACHE |
| Title : Cloning and expression of acetylcholinesterase from Bungarus fasciatus venom. A new type of cooh-terminal domain\; involvement of a positively charged residue in the peripheral site - Cousin_1996_J.Biol.Chem_271_15099 |
| Author(s) : Cousin X , Bon S , Duval N , Massoulie J , Bon C |
| Ref : Journal of Biological Chemistry , 271 :15099 , 1996 |
| Abstract : Cousin_1996_J.Biol.Chem_271_15099 |
| ESTHER : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedSearch : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedID: 8662867 |
| Gene_locus related to this paper: bunfa-ACHE |
| Title : Contribution of aromatic moieties of tyrosine 133 and of the anionic subsite tryptophan 86 to catalytic efficiency and allosteric modulation of acetylcholinesterase - Ordentlich_1995_J.Biol.Chem_270_2082 |
| Author(s) : Ordentlich A , Barak D , Kronman C , Ariel N , Segall Y , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 270 :2082 , 1995 |
| Abstract : Ordentlich_1995_J.Biol.Chem_270_2082 |
| ESTHER : Ordentlich_1995_J.Biol.Chem_270_2082 |
| PubMedSearch : Ordentlich_1995_J.Biol.Chem_270_2082 |
| PubMedID: 7836436 |
| Title : Differential effects of peripheral site ligands on Torpedo and chicken acetylcholinesterase - Eichler_1994_Mol.Pharmacol_45_335 |
| Author(s) : Eichler J , Anselmet A , Sussman JL , Massoulie J , Silman I |
| Ref : Molecular Pharmacology , 45 :335 , 1994 |
| Abstract : Eichler_1994_Mol.Pharmacol_45_335 |
| ESTHER : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedSearch : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedID: 8114681 |
| Title : Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core - Barak_1994_J.Biol.Chem_269_6296 |
| Author(s) : Barak D , Kronman C , Ordentlich A , Ariel N , Bromberg A , Marcus D , Lazar A , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 269 :6296 , 1994 |
| Abstract : Barak_1994_J.Biol.Chem_269_6296 |
| ESTHER : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedSearch : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedID: 8119978 |
| Title : Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase - Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| Author(s) : Harel M , Schalk I , Ehret-Sabatier L , Bouet F , Goeldner M , Hirth C , Axelsen PH , Silman I , Sussman JL |
| Ref : Proceedings of the National Academy of Sciences of the United States of America , 90 :9031 , 1993 |
| Abstract : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| ESTHER : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| PubMedSearch : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| PubMedID: 8415649 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Substrate inhibition of acetylcholinesterase: residues affecting signal transduction from the surface to the catalytic center - Shafferman_1992_EMBO.J_11_3561 |
| Author(s) : Shafferman A , Velan B , Ordentlich A , Kronman C , Grosfeld H , Leitner M , Flashner Y , Cohen S , Barak D , Ariel N |
| Ref : EMBO Journal , 11 :3561 , 1992 |
| Abstract : Shafferman_1992_EMBO.J_11_3561 |
| ESTHER : Shafferman_1992_EMBO.J_11_3561 |
| PubMedSearch : Shafferman_1992_EMBO.J_11_3561 |
| PubMedID: 1396557 |
| Title : Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. - Lockridge_1990_Pharmacol.Ther_47_35 |
| Author(s) : Lockridge O |
| Ref : Pharmacol Ther , 47 :35 , 1990 |
| Abstract : Lockridge_1990_Pharmacol.Ther_47_35 |
| ESTHER : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedSearch : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedID: 2195556 |
| Gene_locus related to this paper: human-BCHE |
| Title : Ligand stabilization of cholinesterases - Payne_1989_Biochim.Biophys.Acta_999_46 |
| Author(s) : Payne CS , Saeed M , Wolfe AD |
| Ref : Biochimica & Biophysica Acta , 999 :46 , 1989 |
| Abstract : Payne_1989_Biochim.Biophys.Acta_999_46 |
| ESTHER : Payne_1989_Biochim.Biophys.Acta_999_46 |
| PubMedSearch : Payne_1989_Biochim.Biophys.Acta_999_46 |
| PubMedID: 2804138 |
| Title : Structure-activity relationship of reversible cholinesterase inhibitors including paraquat - Seto_1988_Arch.Toxicol_62_37 |
| Author(s) : Seto Y , Shinohara T |
| Ref : Archives of Toxicology , 62 :37 , 1988 |
| Abstract : Seto_1988_Arch.Toxicol_62_37 |
| ESTHER : Seto_1988_Arch.Toxicol_62_37 |
| PubMedSearch : Seto_1988_Arch.Toxicol_62_37 |
| PubMedID: 3190453 |
| Title : Differential inhibition of plasma cholinesterase variants using the dibutyrate analogue of pancuronium bromide - Whittaker_1981_Hum.Hered_31_242 |
| Author(s) : Whittaker M , Britten JJ |
| Ref : Hum Hered , 31 :242 , 1981 |
| Abstract : Whittaker_1981_Hum.Hered_31_242 |
| ESTHER : Whittaker_1981_Hum.Hered_31_242 |
| PubMedSearch : Whittaker_1981_Hum.Hered_31_242 |
| PubMedID: 7287015 |
| Title : Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding - Taylor_1975_Biochemistry_14_1989 |
| Author(s) : Taylor P , Lappi S |
| Ref : Biochemistry , 14 :1989 , 1975 |
| Abstract : Taylor_1975_Biochemistry_14_1989 |
| ESTHER : Taylor_1975_Biochemistry_14_1989 |
| PubMedSearch : Taylor_1975_Biochemistry_14_1989 |
| PubMedID: 1125207 |